Documento de consenso de GeSIDA/Plan Nacional sobre el SIDA respecto al tratamiento antirretroviral en adultos infectados por el virus de la inmunodeficiencia humana
ACTUALIZACIÓN 2O22
Notas de la Versión:
Marcado en amarillo el contenido actualizado.
COMITÉ DE REDACCIÓN Y AGRADECIMIENTOS
Comité de redacción*
Coordinadores/as
Redactores/as Generales
Redactores/as y Revisores/as
Agradecimientos
Comité de redacción*
Coordinadores/as
Esteban Martínez (GeSIDA) Hospital Clinic Universitari / IDIBAPS. UB. Barcelona.
José Ramón Arribas (GeSIDA) Hospital Universitario La Paz. IdiPAZ. Madrid.
Rosa Polo (PNS) DCVIHT. Plan Nacional sobre el Sida. MS. Madrid.
Redactores/as Generales
Juan González García Hospital Universitario La Paz. IdiPAZ. Madrid
Rosario Palacios Hospital Universitario Virgen de la Victoria. IBIMA. Málaga.
Redactores/as y Revisores/as
Antonio Antela (GeSIDA) Complejo Hospitalario Universitario de Santiago. Santiago de Compostela.
Juan Ambrosioni (GeSIDA) Hospital Clinic Universitari. IDIBAPS. Universidad de Barcelona. Barcelona.
Victor Asensi (GeSIDA) Hospital Central de Asturias. Universidad de Oviedo. Oviedo.
Enrique Bernal (GeSIDA) Hospital General Universitario Reina Sofía. Murcia.
José Luis Blanco (GeSIDA) Hospital Clinic Universitari. IDIBAPS. Universidad de Barcelona. Barcelona.
José Ramón Blanco (PNS) Hospital San Pedro-CIBIR. Logroño.
Alfonso Cabello (PNS) Hospital Fundación Jiménez Díaz. Madrid.
José Luis Casado (GeSIDA) Hospital Ramón y Cajal. IRYCIS. Madrid.
Carmen de Mendoza (GeSIDA) Hospital Universitario Puerta de Hierro/IDIPHIM. Madrid.
Carlos Dueñas (PNS) Hospital Clínico Universitario de Valladolid.
Ana González-Cordón (GeSIDA) Hospital Clinic Universitari. IDIBAPS. UB. Barcelona.
Carmen Hidalgo (PNS) Hospital Universitario Virgen de las Nieves. Granada.
Juan Emilio Losa (PNS) Hospital Universitario Fundación Alcorcón. Madrid.
Josep Mallolas (GeSIDA) Hospital Clinic Universitari. IDIBAPS. Universidad de Barcelona. Barcelona.
Ana Mariño (PNS) Mar Masià (GeSIDA) Complexo Hospitalario Universitario de Ferrol. El Ferrol.
Mar Masià (GeSIDA) Hospital General Universitario de Elche. Universidad Miguel Hernández. Alicante.
Celia Miralles (PNS) Hospital Álvaro Cunqueiro. Vigo.
Rocío Montejano (GeSIDA) Hospital Universitario La Paz. IdiPAZ. Madrid.
Marta Montero (PNS) Hospital Universitario La Fe. Valencia.
María Luisa Montes (GeSIDA) Hospital Universitario La Paz. IdiPAZ. Madrid.
Roger Paredes (GeSIDA) Hospital Germans Trias i Pujol. IGTP. Badalona.
Jose Antonio Pérez Molina (GeSIDA) Hospital Ramón y Cajal. IRYCIS. Madrid.
Jose Antonio Pineda (GeSIDA) Hospital Universitario de Valme. Sevilla.
Daniel Podzamczer (GeSIDA) Hospital Universitari de Bellvitge. IDIBELL. L’Hospitaletde Llobregat. Barcelona.
Eva Poveda (GeSIDA) Complexo Hospitalario Universitario de Vigo. IISGS. Vigo.
Federico Pulido (GeSIDA) Hospital Universitario 12 Octubre-Instituto de Investigación 1+12. Madrid.
Antonio Rivero (GeSIDA) Jesús Santos (GeSIDA) Hospital Universitario Reina Sofía. IMIBIC. Córdoba.
Jesús Santos (GeSIDA)Hospital Universitario Virgen de la Victoria. IBIMA. Málaga.
Jesús Sanz Sanz (PNS) Hospital Universitario de la Princesa. IIS La Princesa. Madrid.
Sergio Serrano (PNS) Hospital Ramón y Cajal. IRYCIS. Madrid.
Inés Suárez (GeSIDA) Hospital Infanta Sofía. Madrid.
Miguel Torralba (GeSIDA) Hospital Universitario de Guadalajara. Guadalajara.
Jesús Troya (GeSIDA) Hospital Universitario Infanta Leonor. Universidad Complutense. Madrid.
Montserrat Tuset (GeSIDA) Hospital Clínic Universitari. IDIBAPS. Barcelona.
María Velasco (GeSIDA) Hospital Universitario Fundación Alcorcón. Madrid.
Isabel Viciana (GeSIDA) Hospital Universitario Virgen de la Victoria. Málaga.
María J. Vivancos (GeSIDA) Hospital Ramón y Cajal. IRYCIS. Madrid.
Miguel A. von Wichmann (PNS) Hospital Universitario Donostia. Instituto Biodonostia. San Sebastián.
Agradecimientos
La Junta Directiva de GeSIDA y el Plan Nacional sobre el Sida agradecen las aportaciones y opiniones para mejorar el texto de: Hortensia Alvarez, Mª Jesús Pérez Elías, Jara Llenas García, Rafael Rubio García, Pablo Bachiller Luque, del departamento médico de Gilead Sciences (enviado por Raquel Garijo Olmo), del equipo médico MSD-España (enviado por Pedro Ferrer, Itxaso Aguirregabiria, Manuel Cotarelo, Inmaculada Clotet, Fernando Chacón, Oscar Rincón, Nuria Sánchez y Enrique Vacas ), del departamento mádico de ViiV Healthcare (enviado por Jose E Martín Herrero, Beatriz Novoa y Silvia Esteban), del departamento médico de la unidad de enfermedades infecciosas de Janssen España (enviado por Ana Cáceres Núñez) y del departamento médico de Theratechnologies (enviado por Macarena Sierra García).
LISTADO DE ABREVIATURAS
1. INTRODUCCIÓN
Justificación, objetivo y alcance
El uso de los fármacos antirretrovirales (FAR) ha adquirido gran complejidad por la aparición de seis familias, incluyendo más de 40 fármacos y combinaciones, y por sus diferentes características en cuanto a eficacia, toxicidad, resistencias, barrera genética, tropismo, interacciones y uso en situaciones clínicas especiales. Esta complejidad hace necesaria la elaboración y actualización frecuente de guías y recomendaciones sobre el tratamiento antirretroviral (TAR).
El Grupo de Estudio de SIDA (GeSIDA) de la Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC) y el Plan Nacional sobre el SIDA (PNS) editan anualmente un documento de consenso sobre el TAR en adultos.
El objetivo de este documento es transmitir el estado actual del conocimiento sobre el TAR a los profesionales que tratan a adultos con infección por el VIH-1 y proporcionarles recomendaciones que puedan guiar sus decisiones terapéuticas.
GeSIDA y el PNS, junto con otras sociedades científicas elaboran otras recomendaciones referentes a la infección por el VIH donde se incluyen aspectos específicos del TAR. En este documento estos aspectos se tratan de forma somera y se remite al lector a las publicaciones específicas.
Metodología
El panel redactor del documento está integrado por clínicos expertos en la infección por el VIH y el TAR, distribuidos por grupos encargados de actualizar cada sección del documento. Tres miembros del panel actúan como coordinadores y otros dos como redactores generales. Cada grupo revisa los datos más relevantes de las publicaciones científicas y comunicaciones a congresos más recientes, en el caso de esta actualización hasta el 30 de noviembre de 2021, elaboran el texto de cada sección y generan preguntas sobre aspectos no suficientemente consensuados que se someten a votación de todo el panel. El borrador del documento se discute y consensua en una reunión presencial del panel y su redacción provisional se expone durante 15 días en las páginas web de GeSIDA y del PNS para que profesionales, pacientes o quien esté interesado pueda hacer sugerencias que, si procede, son integradas en el documento final.
Cada recomendación de estas guías se califica con una letra que indica su fuerza [A (debe ofrecerse siempre), B (en general debe ofrecerse) o C (debe ofrecerse opcionalmente)] y un número que expresa las pruebas que sustentan dicha recomendación [I (resultados de uno o más ensayos clínicos aleatorizados de aspectos clínicos o de laboratorio o de un metaanálisis), II (de uno o más ensayos no aleatorizados o datos observacionales de cohortes) y III (opinión de expertos)].
2. EVALUACIÓN CLÍNICA Y DE LABORATORIO PARA GUIAR EL TAR
Se aconseja la lectura del Documento de consenso de GeSIDA sobre control y monitorización de la infección por el VIH1.
En este capítulo se recogen las recomendaciones generales y los aspectos relacionados con el uso o la elección del TAR.
Evaluación clínica
La evaluación clínica inicial es una ocasión muy importante para realizar estudio de contactos2, establecer una relación médico-paciente efectiva y duradera y aconsejar hábitos saludables para evitar la transmisión del VIH. En la Tabla 1 se muestran las exploraciones complementarias que deben realizarse en la evaluación inicial y durante el seguimiento.
Recomendaciones
- Realizar al inicio del seguimiento una anamnesis detallada y un examen físico completo que se repetirán como mínimo anualmente (A-III).
- Realizar un estudio de contactos en todos los nuevos diagnósticos con consentimiento del caso índice y garantizando la confidencialidad (A-III).
Evaluación de laboratorio y otros estudios complementarios
Recomendaciones
- En la visita inicial se recomienda realizar una serología del VIH-1/2 en todos los casos en los que no se haya confirmado la infección por el VIH y/o la carga viral palsmática (CVP) sea indetectable (A-III).
- La evaluación inicial de laboratorio debe incluir: hemograma, bioquímica general, serologías y pruebas específicas (véase texto subsiguiente y Tabla 1) (A-II).
2.2.1. LINFOCITOS CD4+
La cifra de linfocitos CD4+ en sangre es el indicador fundamental del estado inmunológico y su determinación es útil para la elección del TAR de inicio y para valorar la respuesta al TAR1. Normalmente se utiliza la cifra absoluta, pero también puede usarse su porcentaje, que es más estable, particularmente en pacientes con leucopenia. Sólo se deben considerar como significativas las variaciones superiores al 30% de las cifras absolutas y del 3% en los valores porcentuales. El valor que aporta su determinación a las decisiones clínicas es menor en pacientes en supresión virológica con una buena reconstitución inmunológica13.
Recomendaciones
- Se debe determinar la cifra absoluta y el porcentaje de linfocitos CD4+ antes de iniciar el TAR y, una vez iniciado cada 3-12 meses, como parámetro de monitorización periódica de la respuesta inmunológica al mismo (A-III).
2.2.2. LINFOCITOS CD8+ Y COCIENTE CD4/CD8
Las elevaciones persistentes de las cifras de linfocitos CD8+ (>1500/uL) o los valores bajos del cociente CD4/CD8 traducen inmunosenescencia4 y se han asociado independientemente a morbimortalidad por complicaciones definitorias de SIDA y no definitorias de SIDA45. Por tanto, su monitorización puede ser de utilidad para identificar a los sujetos que se beneficiarían de un seguimiento más estrecho.
Recomendación
- Se recomienda determinar la cifra de linfocitos CD8+ o el cociente CD4/CD8 cuando se determine la cifra de linfocitos CD4+ en sangre (A-II).
2.2.3. CARGA VIRAL PLASMÁTICA DEL VIH-1
La CVP es el parámetro fundamental para monitorizar la respuesta al TAR y puede ser de utilidad en la elección de este. El objetivo del TAR es conseguir de forma permanente la supresión viral por debajo de 50 cop/mL, lo que evita la transmisión del VIH (indetectable = intransmisible), se asocia a la máxima recuperación inmunológica y previene la aparición de mutaciones de resistencia (MR)1 .
La monitorización de la CVP antes de iniciar el TAR y durante el mismo se ha mostrado una herramienta eficaz para estimar y reforzar la adherencia6. En pacientes con CVP indetectable a los que se modifique el TAR es aconsejable determinar la CVP a las 4-8 semanas del cambio para comprobar que se mantiene la supresión virológica3.
Recomendaciones
- Se debe determinar la CVP antes del inicio del TAR y/o cuando se cambie éste (A-I).
- Se recomienda monitorizar la CVP a las 3-6 semanas del inicio del TAR y posteriormente cada 3-6 meses. Aunque de forma general en pacientes bien controlados con TAR la determinación de CVP debe de ser cada 6 meses, en pacientes clínicamente estables, con CVP repetidamente suprimida y cifras de linfocitos CD4+ >300 células/μL, este intervalo de tiempo puede alargarse incluso hasta 12 meses (A-II).
- Se debe determinar la CVP con una técnica con un límite de detección de CVP de al menos 50 cop/mL para confirmar y monitorizar la supresión virológica (A-I).
2.2.4. RESISTENCIA GENOTÍPICA DEL VIH-1 FRENTE A FÁRMACOS ANTIRRETROVIRALES
La utilidad clínica de las resistencias a antirretrovirales en España es motivo de un documento de GeSIDA que se recomienda consultar7. La prevalencia estimada en España de resistencias primarias en los genes de la transcriptasa inversa y la proteasa es del 7,9%78. La prevalencia de MR que afectan a los inhibidores de transcriptasa inversa análogos de nucleósidos (ITIAN) e inhibidores de proteasa (IP) recomendados para el TAR de inicio es excepcional, siendo mayor para los inhibidores de transcriptasa inversa no nucleósido/tido (ITINN)8 (con la excepción de DOR). La transmisión de resistencias en el gen de la integrasa en la actualidad es inferior al 0,5%8. En España no disponemos de datos específicos de pacientes con historia de uso de profilaxis pre-exposición (PrEP) en el período peri-infección por el VIH, pero algunas experiencias han reportado prevalencias de la MR M184V de al menos el 25%9 por lo que en estos pacientes podría suponer un riesgo de fracaso iniciar biterapia con DTG+3TC o con TDF/3TC/DOR.
Recomendaciones
- Siempre que sea posible se recomienda realizar un estudio genotípico de resistencias del VIH-1 en la transcriptasa inversa y en la proteasa en todos los pacientes antes de iniciar TAR, ampliando a la integrasa si hay evidencia de transmisión a partir de un paciente tratado con INI (A-II).
- Se recomienda esperar a conocer el resultado del estudio genotípico de resistencias si se va a iniciar el TAR con una pauta basada en ITINN para los que existe mayor prevalencia de resistencias transmitidas (EFV, NVP y RPV) (A-II) y cuando se vaya a iniciar con DTG+3TC o con TDF o TAF+XTC+DOR en un paciente con historia de uso de PrEP (A-III). En los demás casos, revisar la prueba en cuanto esté disponible especialmente si se ha utilizado una pauta de baja barrera genética (A-III).
- Se recomienda la realización de un estudio genotípico de resistencias del VIH- 1 en todos los pacientes con fracaso virológico (FV), incluyendo resistencias en la integrasa si el régimen incluye un INI (A-I).
2.2.5. DETERMINACIÓN DEL ALELO HLA-B*5701
Los portadores del alelo HLA-B*5701 tienen un riesgo aproximado de presentar una reacción de hipersensibilidad (RHS) a ABC del 50%10.
Recomendaciones
- Se recomienda determinar el HLA-B*5701 en todos los pacientes antes de iniciar un régimen de TAR que contenga ABC (A-I).
- No se debe prescribir ABC si la prueba del HLA-B*5701 es positiva (A-I).
2.2.6. DETERMINACIÓN DEL TROPISMO DEL VIH-1
Maraviroc (MVC) es un FAR antagonista del correceptor CCR5 que se debe prescribir exclusivamente en los pacientes con infección por cepas del VIH-1 que sean R5- trópicas. El tropismo del VIH-1 se analiza con métodos genotípicos, que determinan la secuencia de la región V3 de la envuelta viral (gp120)11.
Recomendaciones
- Se debe determinar el tropismo del VIH-1 cuando se vaya a utilizar un antagonista del receptor CCR5 (A-I) y cuando fracase un régimen con un antagonista del receptor CCR5 (A-I).
Tablas:
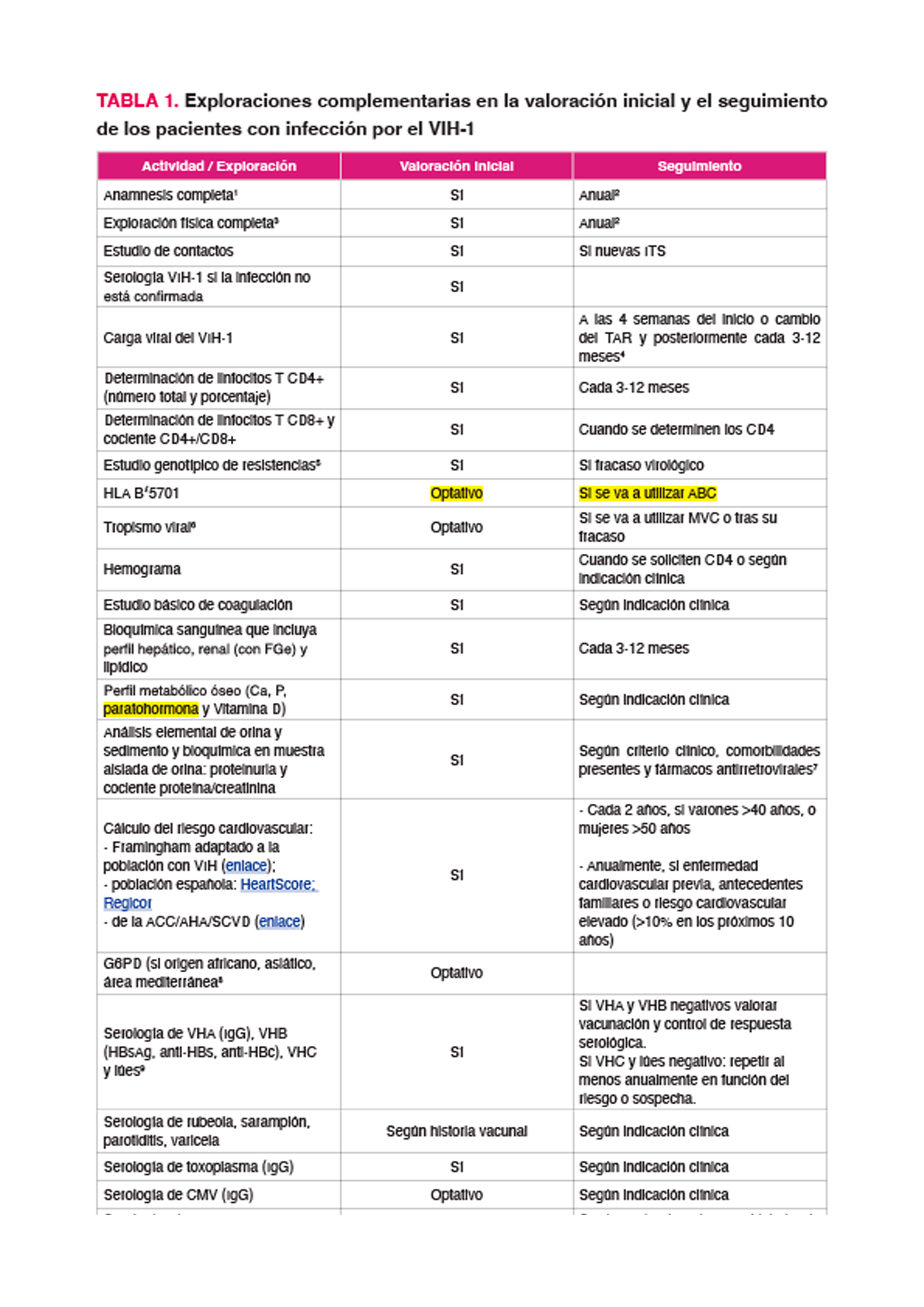
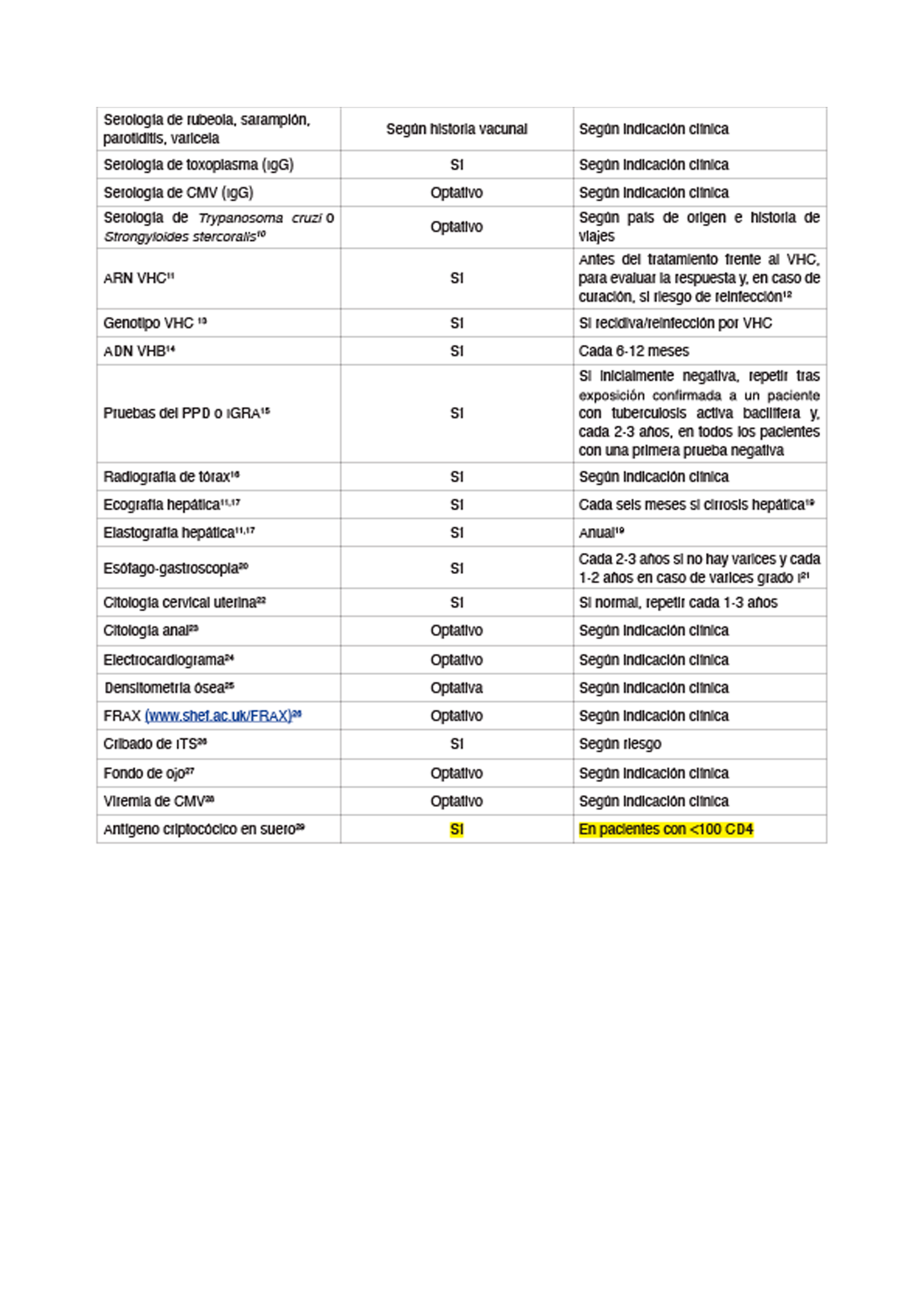
Nota: estas recomendaciones deben considerarse orientativas y pueden ser modificadas de acuerdo con el juicio clínico de los profesionales responsables de la atención al paciente.
1 Debe incluir historia vacunal, uso de fármacos, hábitos sexuales y consumo de tóxicos.
2 O siempre que el paciente lo requiera.
3 Debe incluir medida de la presión arterial, peso, talla y medidas antropométricas (perímetro de cintura).
4 Se puede considerar determinar la CVP y los linfocitos T CD4+ con menos frecuencia (cada 6-12 meses) en pacientes clínicamente estables, con CVP repetidamente suprimida y cifras de linfocitos T CD4+ repetidamente > 300 células/μL (según criterio médico). Consultar el Documento de consenso de GeSIDA sobre control y monitorización de la infección por el VIH de 2018.
5 Debe incluir la transcriptasa inversa y la proteasa; la integrasa sólo si hay evidencia de transmisión a partir de un paciente tratado con INI o el paciente fracasa durante un régimen que incluya esta familia.
6 Realizar solo si se prevé utilizar MVC en el esquema terapéutico.
7 Si se usa TDF o fármacos nefrotóxicos, realizar a 1 y 3 meses de iniciado el tratamiento y posteriormente cada 6 meses. Si diabetes mellitus o hipertensión arterial, determinar además microalbuminuria y cociente albúmina/creatinina en muestra aislada de orina.
8 En caso de iniciar profilaxis con dapsona o sulfonamida.
9 Inicialmente test treponémico y no treponémico, posteriormente únicamente no treponémico si el treponémico es positivo.
10 Trypanosoma y Strongyloides en personas procedentes de áreas con alta prevalencia de infestación, sobre todo si se sospecha la misma (p.e. eosinofilia en la estrogiloidosis). En las personas inmigrantes se debe considerar la realización de serologías según las recomendaciones de evaluación y vacunación de enfermedades prevenibles indicadas para esta población.
11 Si infección por VHC. Se recomienda consultar las Guías EASL: https://easl.eu/publications/clinical- practice-guidelines/
12 Cada 6-12 meses en HSH o “chemsex”.
13 Solo si infección activa por VHC (RNA+).
14 Si co-infección por VHB se recomienda su determinación antes de iniciar el TAR. Recomendable en pacientes con antiCore aislado.
15 La sensibilidad del PPD es menor en personas con infección por el VIH y se acentúa a mayor inmunosupresión, recomendándose la determinación de IGRA si la prueba de PPD ha sido negativa, especialmente en pacientes muy inmunosuprimidos (<200 CD4). La especificidad de PPD disminuye en vacunados (BCG), por lo que se recomienda la utilización de IGRA.
16 Especialmente en pacientes pertenecientes a poblaciones con una elevada prevalencia de tuberculosis, criterios de bronquitis crónica o tabaquismo.
17 Si co-infección por VHB.
18 En VHC Fibroscan > 14kpa y en VHB > 9 kpa con ALT normal y > 12 kpa con ALT elevada.
19 Salvo nuevas hepatopatías, sólo 1-2 controles tras RVS de VHC.
20 Si cirrosis hepática.
21 Falta evidencia sobre la periodicidad en VHC tras RVS.
22 Si se detectan células atípicas, realizar colposcopia y biopsia.
23 Inicialmente, ofrecer a los pacientes con hábitos de riesgo (HSH y mujeres que practican coito anal receptivo) y en los que presentan lesiones perianales o genitales secundarias a VPH. Si citología anormal, debe realizarse anuscopia de alta resolución y biopsia (el tratamiento de la displasia anal de alto grado ha demostrado disminuir la incidencia de cáncer anal en el estudio ANCHOR).
24 Especialmente en pacientes con factores de riesgo cardiovascular y/o que vayan a iniciar fármacos que puedan producir problemas de conducción cardíaca.
25 Se deben de identificar los factores de riesgo de desarrollar alteraciones de la densidad mineral ósea. Se recomienda seguir las recomendaciones el Documento de consenso de GeSida sobre osteoporosis en la infección por VIH de mayo de 2016 (enlace).
26 Valorar el riesgo de ITS y hacer cribado de las mismas siguiendo las recomendaciones del Documento de Consenso sobre las Infecciones de transmisión sexual en adultos, niños y adolescentes de GeSIDA/PNS/ GEITS/SEIP, 2017 (enlace).
27 Considerar en pacientes con <50 CD4, en los que la retinitis por CMV puede cursar de manera asintomática inicialmente hasta en un 50% de los casos.
28 El riesgo de enfermedad de órgano diana por CMV aumenta drásticamente en pacientes con <100 CD4. En pacientes asintomáticos de riesgo (<100 CD4 y viremia de CMV) el tratamiento con valganciclovir no ha demostrado beneficio clínico, por lo que la determinación debe realizarse si hay sospecha de enfermedad por CMV. (enlace)
29 El tratamiento anticipado con fluconazol ha demostrado disminuir la mortalidad en África (enlace), aunque esta intervención no se ha estudiado en Europa, y su positividad se ha relacionado con mayor mortalidad en España (enlace).
Bibliografía:
Consenso de GeSIDA sobre control y monitorización de la infección por el (Actualización abril 2018). http://gesida-seimc.org/wp-content/uploads/2018/02/gesida_DC_Control_Monitorizacion_VIH.pdf (Consultado el 30/10/2021)
Ferreira A, Young T, Mathews C, Zunza M, Low N. Strategies for partner notification for sexually transmitted infections, including Cochrane Database Syst Rev 2013:CD002843.
Caniglia EC, Sabin C, Robins JM, et When to monitor CD4 cell count and HIV RNA to reduce mortality and AIDS-defining illness in virologically suppressed HIV-positive persons on antiretroviral therapy in high-income countries: a prospective observational study. J Acquir Immune Defic Syndr 2016; 72:214-21.
Serrano-Villar S, Sainz T, Lee S a., et al. HIV-infected individuals with low CD4/CD8 ratio despite effective antiretroviral therapy exhibit altered T cell subsets, heightened CD8+ T cell activation, and increased risk of non-AIDS morbidity and mortality. PLoS Pathog 2014; 10:e1004078.
Mussini C, Lorenzini P, Cozzi-Lepri A, et al. CD4/CD8 ratio normalisation and non-AIDS related events in individuals with HIV who achieve viral load suppression with antiretroviral therapy : an observational cohort Lancet HIV 2015; 2:e98-106.
Bonner K, Mezochow A, Roberts T, et al. Viral load monitoring as a tool to reinforce adherence: a systematic J Acquir Immune Defic Syndr 2013; 64:74-8.
Grupo de Estudio de Sida de la Sociedad Española de Enfermedades infecciosas. Documento sobre la utilidad clínica de las resistencias a antirretrovirales. Octubre 2018. http://gesida-seimc.org/wp-content/uploads/2018/09/gesida_documento_sobre_la_utilidad_clinica_de_las_resistencias_a_antirretrovirales.pdf (Consultado el 30/10/2021)
Alvarez M, Casas P, de Salazar A, et al. Surveillance of transmitted drug resistance to integrase inhibitors in Spain: implications for clinical practice. J Antimicrob Chemother. 2019 Jun 1;74:1693-1700. doi: 1093/jac/dkz067.
Johnson KA, Chen MJ, Kohn R, et Acute HIV at the time of initiation of Pre-exposure or Post-exposure Prophylaxis: impact on drug resistance and clinical outcomes. J Acquir Immune Defic Syndr (1999) 2021; 87:818-25.
Hughes CA, Foisy MM, Dewhurst N, et al. Abacavir hypersensitivity reaction: an Ann Pharmacother 2008; 42:387-96.
Poveda E, Alcamí J, Paredes R, et Genotypic determination of HIV tropism - clinical and methodological recommendations to guide the therapeutic use of CCR5 antagonists. AIDS Rev 2010;12:135-48.
3. TRATAMIENTO ANTIRRETROVIRAL INICIAL
Los objetivos del TAR son conseguir la máxima y más duradera supresión de la CVP, restablecer y preservar la función inmunológica, reducir la morbilidad asociada a la replicación del VIH-1 y su efecto sobre otras comorbilidades, aumentar la supervivencia y prevenir la transmisión del VIH-1. Aunque el TAR debe iniciarse tan pronto como sea posible, es importante valorar de forma individualizada el momento más adecuado de inicio del TAR y los FAR que deben formar parte del régimen inicial. La situación clínica del paciente, así como su disposición y motivación son factores críticos a la hora de decidir el momento parainiciar el TAR.
Cuándo iniciar el TAR
El TAR debe iniciarse en todos los pacientes con infección por el VIH-1, con o sin sintomatología, y con independencia del número de linfocitos T CD4+ Tabla 2. Como excepción se consideran los pacientesque mantienen CVP indetectable de forma mantenida sin TAR (controladores de élite). En este caso noexiste información suficiente que permita valorar el efecto beneficioso del TAR, por lo que no se puedeestablecer una recomendación al respecto.
La recomendación de inicio en todos los pacientes, con la excepción mencionada se sustenta sobre todoen dos grandes ensayos clínicos aleatorizados12.
En el ensayo START (Strategic Timing of AntiRetroviral Treatment)1, se incluyeron 4.685 personas infectadas por el VIH-1 con una cifra de linfocitos CD4+ >500 células/μL, y fue- ron seguidas durante untiempo medio de 3 años. Se aleatorizaron a iniciar TAR de forma inmediata o a diferirlo hasta que el recuento de linfocitos CD4+ fuera <350 células/μL.
La variable principal fue la proporción de pacientes que presentaban un evento definitorio de SIDA, unacomplicación grave no asociada a SIDA o muerte por cualquier motivo. Este criterio ocurrió en el 1,8% de los pacientes que iniciaron TAR de forma inmediata y en el 4,1% de los que lo difirieron (una reducción deriesgo del 57% [IC95%: 38% a 70%]). Esta diferencia fue considerada suficientemente importante yconsistente como para interrum- pir el estudio y recomendar TAR a todos los pacientes que aún no lorecibían.
El estudio TEMPRANO2 se llevó a cabo en Costa de Marfil. Se incluyeron 2.056 pacientes sin TAR previo y una cifra de linfocitos CD4+ inferior a 800 céulas/μL que se asignaron aleatoriamente a recibir TAR de forma inmediata o a diferirlo hasta presentar criterios de tratamiento de acuerdo con lasrecomendaciones de la OMS vigentes en cada momento. La variable principal fue el desarrollo de SIDA, de cáncer no asociado a SIDA, de enfer- medad bacteriana invasiva, o muerte por cualquier causa en un periodo de 30 meses.
La mediana de linfocitos CD4+ en el momento de la inclusión fue de 460 células/μL. El inicio inmediato deTAR se relacionó con una disminución de eventos principales del 44% (IC95%: 24 a 59%). Cuando seanalizaron por separado los pacientes que entraron en el estudio con una cifra de linfocitos CD4+ superiora 500 células/μL (n=849), el inicio inme- diato se asoció con un descenso del riesgo de presentar alguno delos eventos primarios en los 30 meses siguientes del 44% (IC95%: 6 a 67%), a pesar de que durante elestudio un 41% de los pacientes asignados a diferir el TAR lo iniciaron igualmente, y la mediana delinfocitos CD4+ no fue en ningún momento del seguimiento inferior a 500 céulas/μL. Al igual que en elestudio START, la mayoría de los eventos en este subgrupo se produjeron con recuentos de linfocitosCD4+ superiores a 500 células/μL.
Por otra parte, se ha demostrado que el inicio del TAR se asocia con un menor riesgo de transmisión delVIH-1 y reducción de nuevas infecciones3.
Varios ensayos clínicos realizados en países con recursos económicos limitados4 y algu- nas experiencias observacionales en Londres5, San Diego6 y San Francisco7, han mostrado que el inicio rápido del TAR (el mismo día del diagnóstico o en la primera semana) favorece la retención de los pacientes en la asistencia e incrementa la proporción de pacientes con supresión virológica, lo que podría tener implicaciones de salud pública al disminuir la transmisibilidad.
En el estudio DIAMOND, un ensayo clínico no comparativo realizado en Estados Unidos, se evaluó en 109 pacientes la eficacia de DRV/c/FTC/TAF iniciado en los primeros 14 días tras el diagnóstico, antes dedisponer de las determinaciones basales de laboratorio. Este tratamiento mostró una eficacia elevada conCVP <50 cop/mL en el 84% de los pacientes a las 48 semanas en el análisis snapshot y 96% en elanálisis de datos observados (n=96), sin discontinuaciones por fracaso8. Por otro lado, en el estudio STAT9, 131 pacientes sin tratamiento previo fueron incluidos en un estudio de brazo único, todos los cuales recibieronla combinación de DTG/3TC en los primeros 14 días tras el diagnóstico de infección por VIH-1 sin resultados analíticos previos. A lo largo de las primeras 24 semanas, el tratamiento se modificó en 8 pacientes (5 con infección por VHB, uno con la mutación M184V al inicio, uno por evento adverso y uno por decisión del paciente). A las 24 semanas, endpoint primario, el 78% estaban con CVP <50 cop/mL (snapshot). A las 48 semanas, el porcentaje de pacientes con CVP <50 cop/mL fue del 82%10. Es de destacar que en los escasos fracasos virológicos, no se detectó emergencia de resistencias. En el estudioFAST, desarrollado en Francia y presentado en la Conferencia de la EACS de 202111, se evaluó la combinación BIC/FTC/TAF en una estrategia de inicio rápido. Se incluyeron 117 participantes. El análisis por intención de tratamiento incluyó a 112, pues 5 fueron incluidos erróneamente. En el análisis por“snapshot”, a las 24 semanas, el 80,4% estaban con CVP <50 cop/mL, 9,8% con CVP >50 cop/mL, 1,8% habían abandonado por evento adverso o muerte y 8% por otras causas. Hubo diferencias en el porcentaje de pacientes con CVP <50 cop/mL a las 24 semanas, según la CVP inicial, siendorespectivamente del 88,7%, 75,9% y 61,9%, según si la CVP inicial era, respectivamente, <100.000cop/mL, entre 100.000 y 500.000 cop/mL o >500.000 cop/mL. No se detectaron mutaciones asociadas a resistencias en los pacientes con CVP detectable a las 24 semanas.
En caso de plantearse el inicio del TAR de forma inmediata, sin disponer de toda la información depruebas complementarias, la selección adecuada de la pauta de TAR es esencial. Debe cumplir: facilidad de toma, buena tolerabilidad, no requerir estudio previo de HLA-B*5701, mínimo riesgo de interacciones farmacológicas, alta probabilidad de mantener actividad antiviral en presencia de CVP elevadas, cifras bajas de linfocitos CD4+ o de virus con mutaciones de resistencia basales e idealmente capacidad de suprimir la replicación del VHB en caso de coinfección por el mismo.
Recomendaciones
- Se recomienda la administración de TAR a todos los pacientes con infección por el VIH-1 paraevitar la progresión de la enfermedad, limitar el efecto nocivo sobre posibles comorbilidades ydisminuir la transmisión del virus (A-I). Como excepción se consideran los pacientes que mantienen CVP indetectable de forma mantenida sin TAR (controladores de élite).
- Se recomienda iniciar el TAR tan pronto como sea posible, idealmente en los primeros 14 díastras el diagnóstico (A-II).
- El inicio del TAR debe valorarse siempre individualmente (A-III). Se debe realizar siempre una determinación de linfocitos CD4+ y CVP previa al inicio del tratamiento, aunque no es imprescindible esperar hasta disponer de los resultados si se utiliza una pauta cuya recomendaciónno esté condicionada a sus valores (A-III). Además, debe prepararse al paciente, proporcionándoleinformación sobre los objetivos del tratamiento y las distintas opciones, seleccionando el esquema terapéutico que mejor se adapte al estilo de vida, comorbilidades, posibles interacciones y valorando el riesgo de mala adherencia (A-III).
Qué combinación de antirretrovirales debe utilizarse
Las pautas recomendadas para el tratamiento inicial de la infección por el VIH-1 en el momento actualconsisten en una combinación de dos o tres fármacos. Las pautas triples deben incluir dos ITIAN asociadosa un INI, a un ITINN o a DRV/p (Tabla 3). La única pauta doble recomendada en el momento actual comoTAR de inicio consiste en la combinación de un ITIAN (3TC) y un INI (DTG).
Con estas combinaciones se puede conseguir una CVP <50 cop/mL a las 48 semanas de tratamiento enmás del 85% de los casos.
En el caso de pacientes embarazadas o de pacientes con tuberculosis estas recomendaciones no sonválidas y se debe utilizar la información existente en los apartados correspondientes de este documento yen las guías específicas.
Recomendación
- El TAR de inicio consiste en una combinación de dos o tres FAR, en alguna de las combinaciones quese detallan en la (Tabla 3) (A-I).
3.2.1. INHIBIDORES DE LA TRANSCRIPTASA INVERSA ANÁLOGOS DENUCLEÓSIDOS
En España se utilizan tres ITIAN: 3TC, FTC y ABC. También se dispone de un análogo denucleótido (TFV). A efectos prácticos, la abreviatura ITIAN en esta guía incluye también a TFV. TFV está comercializado con 2 formulaciones diferentes: tenofovir disoproxilo (TDx) y tenofoviralafenamida (TAF). Todo el desarrollo clínico de TDx se ha realizado utilizando la sal defumarato (TDF), aunque en la actualidad existe como fármaco genérico como otras sales (maleato, fosfato, succinato). Comparado con TDF, TAF requiere dosis menores (10 mg/día cuando se utilizacon antirretrovirales potenciados con RTV o COBI y 25 mg con el resto de antirretrovirales) dado que se concentra selectivamente como fármaco activo (TFV-difosfato) en las células diana y, por tanto, las concentraciones plasmáticas son un 90% menores. TAF ha demostrado una eficacia superior a TDF tras 3 años de seguimiento, ambos en combinación con EVG/c/FTC12. Así mismo, en ensayos clínicos aleatorizados ha demostrado menor toxicidad renal y menor reducción de la densidad mineral ósea que TDF1213. Además, TAF puede utilizarse con filtrados glomerularessuperiores a 30 ml/min, mientras que TDx no está indicado por debajo de 50 ml/min. Un metaanálisisde ensayos clínicos que ha comparado TDF frente a TAF muestra diferencias en toxicidad renal yósea cuando se utilizan en pautas que incluyen potenciadores (RTV o COB). No obstante, no se evidencian diferencias entre ambos agentes cuando se utilizan en pautas no potenciadas14.
Se consideran como combinaciones de ITIAN de elección las formadas por FTC/TAF y por ABC/3TC, que deberían administrase siempre que sea posible en preparados coformulados. La utilización de TFV como TDx puede considerarse una alternativa a TAF en regímenes que no incluyan un potenciador, siempre que se excluya la presencia de alteración renal o de alteración de la densidad mineral ósea y no existan otros factores que aumenten el riesgo de desarrollarlas.
No existe evidencia clínica que permita afirmar diferente eficacia de 3TC y FTC, por lo que el uso de uno u otro ITIAN en los regímenes seleccionados depende fundamentalmente de la experiencia disponible en su uso conjunto con los otros FAR de la combinación.
Combinaciones con FTC/TFV frente a combinaciones con ABC/3TC
Sólo un estudio ha comparado hasta este momento TAF frente a ABC en el TAR de inicio, aunque enregímenes diferentes (BIC/FTC/TAF vs DTG/ABC/3TC)15. En este estudio la eficacia de la combinación con TAF fue no inferior a la de ABC, aunque en el grupo que recibió la pauta con ABC la frecuencia de pacientes que refirieron náuseas fue mayor (10% vs 22%).
Varios estudios han comparado TDF frente a ABC. En el ensayo clínico ACTG 520216 se comparó de forma ciega el inicio de TAR con ABC/3TC o FTC/TDF en 1.857 pacientes. Los participantes fueron aleatorizados además a recibir ATV/r o EFV de forma abierta. Entre los pacientes con CVPbasal igual o mayor de 100.000 cop/mL, tanto el tiempo hasta el FV como el tiempo hasta el primer efecto adverso de grado 3-4 fueron significativamente más cortos en el brazo de ABC/3TC que en el brazo de FTC/TDF. En los pacientes con CVP inferior a 100.000 cop/mL no hubo diferencias en eficacia virológica entre ABC/3TC y FTC/TDF, independientemente de que se administraran conATV/r o EFV1718.
Tres estudios en fase III diseñados para comparar el TAR de inicio con DTG, frente a otros FAR recomendados (EFV en el estudio SINGLE19, RAL en el estudio SPRING-2(20) o DRV/r en el estudio FLAMINGO(21)) han mostrado una eficacia similar de ABC/3TC o FTC/TDF. Sin embargo,dichos ensayos clínicos no permiten establecer comparaciones formales ya que, o bien la elección del ITIAN no fue aleatorizada, quedando a criterio del investigador2021, o las distintas combinaciones deITIAN iban asociadas a un tercer fármaco también distinto19.
Recomendaciones
- Las combinaciones de ITIAN de elección para regímenes de inicio son FTC/ TAF o ABC/3TC(A-I). Se recomienda su uso en coformulación (A-II).
- TDF puede utilizarse como alternativa a TAF en regímenes que no incluyan un potenciador(RTV o COBI), siempre que se excluya la presencia de alteración renal u ósea y no existanotros factores que aumenten el riesgo de desarrollarlas (C-I).
- La combinación ABC/3TC, con un ITINN o un IP/p debe evitarse en pacientes con CVP elevada(>100.000 cop/mL) (A-I).
3.2.2. INHIBIDORES DE LA INTEGRASA
Existen cuatro INI con aprobación para su uso como TAR de inicio: RAL, EVG, DTG y BIC.
Este Panel recomienda de forma preferente 2 INI (BIC y DTG), como parte del TAR de inicio.
Los INI pueden utilizarse en la mayoría de pacientes con una elevada eficacia y un riesgo bajo detoxicidad e interacciones en comparación con otras pautas. Además, se ha observado una mayor rapidez en la supresión virológica con las pautas que incluyen un INI en comparación con otrasfamilias de FAR.
RAL fue el primer INI comercializado. Se puede utilizar en dosis de 400 mg administrado dos veces al día (400 mg BID), o en dosis de 1.200 mg una vez al día (2 comprimidos de 600 mg QD)22.
Combinado con FTC/TDF, RAL 400 mg BID ha demostrado superioridad frente a DRV/r y a ATV/r23y no inferioridad frente a EFV, alcanzando una eficacia superior a éste al cuarto y quinto años deseguimiento24. Administrado en dosis de 1.200 mg QD, RAL ha demostrado una eficacia no inferior y tolerancia similar a 400 mg BID, ambas administradas junto a FTC/TDF22. Aunque no existe evidencia directa del uso de RAL con FTC/TAF, este Panel considera que las ventajas en seguridadmostradas en las comparaciones directas de TAF frente a TDF, junto con otros FAR, en pacientes sin tratamiento previo1225 son suficientes para recomendar RAL asociado a TAF/FTC cuando sedecida su uso como pauta de inicio.
Pese a su eficacia, buena tolerabilidad, seguridad y excelente perfil de interacciones, este Panel considera que RAL debe ser considerado una alternativa debido a su peor perfil de resistencias en el FV, en comparación con los INI de segunda generación (DTG, BIC) y por no poder administrarseen un comprimido único.
EVG no se incluye entre las pautas preferentes ni alternativas debido a su mayor potencial deinteracciones con respecto a los otros INI no potenciados, que pueden también administrarse en combinación con FTC/TAF, y no tener ninguna ventaja sobre las recomendadas.
DTG se administra como un comprimido de 50 mg QD en pacientes sin TAR previo y no necesitapotenciación. Se ha comparado en ensayos clínicos fase III con FAR de las tres familias que en algúnmomento han sido recomendados en el TAR de inicio.
El ensayo clínico aleatorizado y doble ciego SINGLE1926 demostró la superioridad de DTG/3TC/ABC frente a EFV/FTC/TDF a las 48 semanas, que se mantuvo en la semana 144. Laproporción de FV fue similar en ambos brazos de tratamiento, pero la proporción de interrupciones porefectos adversos fue mayor en el brazo de EFV/ FTC/TDF. La proporción de pacientes en faseavanzada (cifra de CD4 < 200 células/ μL) fue baja (14%).
El ensayo clínico aleatorizado ADVANCE realizado en Sudáfrica comparó la eficacia de DTG+FTC/TAF, DTG+FTC/TDF y EFV/FTC/TDF13. El 60% de los pacientes incluidos eran mujeres,el 30% tenía una cifra de CD4+ inicial <200 células/μL y no se disponía de estudio deresistencias basal. A las 48 semanas el porcentaje de pacientes con CVP <50 cop/mL (análisis ITT snapshot) fue 84%, 85% y 79%, respectivamente, demostrándose la no inferioridad de las pautas conDTG respecto a EFV.
Las diferencias en la eficacia entre DTG y EFV se debieron fundamentalmente a un mayor número dediscontinuaciones en el grupo de EFV.
El ensayo clínico aleatorizado abierto NAMSAL ANRS 12313 comparó la eficacia de DTG y EFV 400mg, ambos en combinación con FTC/TDF, en pacientes sin TAR previo27. DTG demostró una eficaciano inferior a EFV 400 mg a las 48 semanas.
DTG ha demostrado eficacia superior al IP/p DRV/r, ambos en combinación con 2 ITIAN, en unensayo clínico aleatorizado abierto (FLAMINGO)2128. Aunque no se observaron diferencias en cuanto a FV, el porcentaje de efectos adversos y discontinuaciones por causas no relacionadas con el fármaco fue mayor en los tratados con DRV/r.
El ensayo clínico aleatorizado abierto ARIA29 en el que se incluyeron solamente mujeres mostró unaeficacia superior de DTG/3TC/ABC frente a ATV/r+FTC/TDF.
Dos ensayos clínicos aleatorizados con el mismo diseño (GEMINI-1 y GEMINI-2)30, han comparado la biterapia DTG+3TC con el TAR triple DTG+TDF/FTC en un total de 1.433 pacientes sin tratamiento previo y con CVP <500.000 cop/mL. DTG+3TC demostró la no inferioridad frente al tratamiento triple,manteniendo una alta eficacia a las 48 semanas (CVP <50 cop/mL 91%, análisis snapshot). La no inferioridad de DTG+3TC se mantuvo en el análisis a las 96 semanas31. En el análisis planificado desubgrupos, no se observaron diferencias en función de la CVP basal mayor o menor de 100.000cop/mL. En cambio, los pacientes que iniciaron DTG+3TC con una cifra basal de CD4+ <200 células/μL mostraron una menor eficacia en el análisis por snapshot, aunque la causa de esta menor eficacia no se debió a un mayor porcentaje de FV, sino a discontinuaciones no relacionadasdirectamente con el tratamiento. Además, el número de pacientes en este subgrupo, comosucede en la mayoría de ensayos clínicos, fue pequeño (n=65, 9%) y no permite extraer conclusionesdefinitivas.
Se presentaron los resultados a 144 semanas en el CROI de 202132, en los que se mantenía la no inferioridad de DTG+3TC, tanto global como en el análisis pormenorizado por subgrupos. Sin embargo, seguía existiendo una menor eficacia en pacientes con una cifra basal de linfocitos CD4+<200 células/μL.
En un meta-análisis en red de 14 ensayos clínicos aleatorizados y doble ciego con pautas de TAR de inicio se ha mostrado que la eficacia y seguridad de DTG+3TC a las 48 semanas de tratamiento es comparable a las combinaciones de TAR triples analizadas, incluso en aquellos pacientes con CVPbasal >100.000 cop/mL33.
Con la información disponible actualmente, el Panel se reafirma en que se necesita disponer de mayor información sobre la eficacia de DTG+3TC en pacientes con una cifra de CD4+ <200 células/μL antes de que pueda recomendarse en este escenario clínico.
Es importante destacar que DTG presenta una barrera a las resistencias superior a RAL y EVG, siendo excepcional la selección de MR tras un FV a una pauta de TAR de inicio con DTG, tanto enasociación con 2 ITIAN como con la biterapia DTG+3TC.
En España no disponemos de datos específicos de pacientes con historia de uso de PrEP en el período de peri-infección por el VIH-1, pero algunas experiencias han comunicado prevalenciaselevadas de la mutación M184V, por lo que en estos pacientes podría suponer un riesgo de fracaso iniciar biterapia con DTG+3TC si no se dispone de estudio de resistencias.
BIC se encuentra disponible en coformulación con FTC y TAF en un único comprimido que seadministra QD (BIC/FTC/TAF).
BIC/FTC/TAF en pacientes sin TAR previo se ha estudiado en dos ensayos clínicos fase III aleatorizados y doble ciego, ambos en comparación con DTG. En el estudio GS-US-380-148915 BIC/FTC/TAF demostró una eficacia no inferior a DTG/3TC/ABC y en el estudio GS-US-380-149034 ,BIC/FTC/TAF se mostró no inferior a DTG+FTC/ TAF. En ambos estudios, BIC/FTC/TAF hamantenido la no inferioridad frente a las pautas con DTG a los 4 años35. El porcentaje de discontinuaciones por efectos adversos fue bajo (2%) y similar a los grupos de tratamiento con DTG. No se observó selección de MR a los fármacos utilizados en ningún paciente.
Recomendaciones
- Se recomiendan como pautas preferentes de TAR de inicio las siguientes combinaciones: BIC/FTC/TAF (A-I); DTG/ABC/3TC o DTG+FTC/TAF (A-I) o DTG+3TC (A-I).
- Se recomienda esperar a conocer el resultado del estudio genotípico de resistencias si se vaa iniciar el TAR con DTG+3TC en un paciente con historia de uso de PrEP (A-III).
- La combinación RAL (400 mg BID o 1200 mg QD) + FTC/TAF (A-III) se considera unaalternativa a las pautas preferentes de TAR de inicio (C-I).
3.2.3 INHIBIDORES DE LA TRANSCRIPTASA INVERSA NO NUCLEÓSIDOS(ITINN)
En España hay cinco ITINN comercializados (NVP, EFV, ETR, RPV y DOR). NVP, EFV y ETR son inductores de algunos isoenzimas del citocromo P450 y, por tanto, pueden interaccionar con otrosfármacos metabolizados por esta vía.
EFV se administra QD (un comprimido de 600 mg, existiendo en presentación coformulada con FTC/TDF en un solo comprimido). Su principal limitación es la aparición frecuente de síntomasrelacionados con el SNC al inicio del tratamiento y que, aunque suelen ser leves y transitorios, pueden ocasionar discontinuaciones del TAR. En distintos análisis de ensayos clínicos independientes se observó un mayor riesgo de ideación y conductas suicidas3637, aunque estaasociación no se ha encontrado en dos estudios de cohortes3839.
RPV se administra QD (un comprimido de 25 mg, existiendo también la presentación coformulada con FTC/TDF o con FTC/TAF en un solo comprimido) siempre con alimentos (al menos 390 Kcal) y está contraindicado su uso con inhibidores de la bomba de protones (IBP).
DOR, se administra QD (un comprimido de 100 mg), existiendo también la presentación coformulada con 3TC/TDF en un solo comprimido. A diferencia de otros NNRTI, no es inductor niinhibidor de CYP3A4 y puede administrarse con IBP.
Actualmente no se recomienda el uso de NVP en pautas de TAR de inicio debido a su mayor riesgo de toxicidad y a no haber demostrado la no inferioridad con respecto a EFV40.
ETR (1 comprimido de 200 mg/12 h), no está aprobada por la EMA para el TAR de inicio.
EFV se ha comparado con IP/p como TAR de inicio, demostrando una eficacia superior a LPV/r41, SQV/r42 o APV/r43. En el estudio ACTG 520218, la eficacia virológica resultó similar en los tratados con ATV/r que en los tratados con EFV independientemente de que se combinaran con ABC/3TC o FTC/TDF. Entre los FV, la emergencia de MR fue significativamente menor con ATV/r. El tiempo hasta el primer evento de seguridad y el primer evento de tolerabilidad fue significativamente más largo para los pacientes con ATV/r cuando la pareja de ITIAN era ABC/3TC, pero no hubo diferencias cuando la pareja de ITIAN era FTC/TDF. Un subanálisis de este estudio mostró unamenor eficacia de ATV/r respecto a EFV en mujeres44.
EFV ha sido comparado con INI en diversos ensayos clínicos aleatorizados de TAR de inicio que han puesto en evidencia una menor eficacia de EFV respecto a RAL (estudio STARTMRK24) y DTG(estudio SINGLE1926). En comparación con EFV 400 mg QD, DTG demostró una eficacia no inferiora las 48 semanas de tratamiento27.
RPV se ha comparado con EFV en tres ensayos clínicos aleatorizados en pacientes sin TAR previo 45464748. Los estudios ECHO45 y THRIVE46 incluyeron pacientes adultos sin TAR previo y sin MR en el estudio genotípico basal. Los participantes fueron aleatorizados a recibir de forma ciega RPV o EFV junto a dos ITIAN (FTC/TDF coformulado en el estudio ECHO y una pareja de ITIAN seleccionada por los investigadores en el estudio THRIVE, que en el 60% de los casos fue también FTC/TDF). Elanálisis combinado de ambos estudios a las 96 semanas demostró la no-inferioridad de RPV con respecto a EFV. La tolerabilidad fue mejor con RPV, con un menor número de discontinuaciones por efectos adversos y sobre todo los relacionados con el SNC. Sin embargo, en el subgrupo depacientes con CVP inicial >100.000 cop/mL, la frecuencia de FV fue superior con RPV (17,6 vs. 7,6%), por lo que no se recomienda el uso de RPV/FTC/TDF en estos pacientes. El FV con RPV se asoció, además, conmayor frecuencia a resistencia genotípica a otros ITINN y a ITIAN (especialmente por la selección delas mutaciones M184I y M184V)47.
En el ensayo clínico STaR48 se compararon de forma abierta las combinaciones en un comprimidoúnico de RPV/FTC/TDF frente a EFV/FTC/TDF como pautas de TAR de inicio. Se demostró la no inferioridad de RPV/FTC/TDF frente a EFV/FTC/TDF tanto a las 48 como a las 96 semanas. En el análisis de subgrupos la eficacia de RPV/FTC/TDF fue superior a EFV/FTC/TDF en los pacientes conCVP basal igual o menor de 100.000 cop/mL, no inferior en los pacientes con CVP >100.000 cop/mLe inferior en pacientes con CVP >500.000 cop/mL. La retirada del tratamiento por efectos adversos,así como la incidencia de efectos adversos neuropsiquiátricos fue menor en los pacientes tratados conRPV en comparación con los tratados con EFV.
No existen ensayos clínicos que hayan comparado RPV con otros FAR en TAR de inicio.
La combinación RPV/FTC/TAF no ha sido evaluada de forma específica como TAR de inicio. Sin embargo, este Panel considera que las ventajas en seguridad mostradas en las comparaciones directas de TAF con TDF en pacientes sin tratamiento previo12, así como en los ensayos clínicos que han comparado la eficacia y seguridad del cambio a RPV/FTC/TAF en pacientes pretratados49son suficientes para recomendar esta combinación como alternativa a las pautas preferentes.
DOR es un nuevo ITINN que se ha estudiado como tratamiento de inicio en ensayos clínicos aleatorizados comparándose con EFV, ambos en coformulación con 3TC/ TDF o con FTC/TDF, respectivamente (Estudio DRIVE-AHEAD)50, y con DRV/r, en combinación con FTC/TDF o 3TC/ABC (Estudio DRIVE-FORWARD)51. En ambos estudios, DOR ha mostrado una eficacia no inferior a sus comparadores en el análisis primario a las 48 semanas. El porcentaje de pacientes con efectos adversos sobre el SNC fue significativamente inferior en los tratados con DOR en comparación conEFV. En un análisis con 96 semanas de seguimiento ciego52, DOR ha demostrado eficacia superiora DRV/r con mejor evolución del perfil lipídico.
Hasta el momento DOR no se ha comparado con INI en ensayos clínicos.
Aunque no existe evidencia directa del uso de DOR con FTC/TAF, este Panel considera que las ventajas en seguridad mostradas en las comparaciones directas de TAF frente a TDF, junto con otros FAR, en pacientes sin tratamiento previo12 son suficientes para recomendar el uso de esta combinación como alternativa a las pautas preferentes.
Recomendaciones
- Actualmente no se considera preferente ninguna pauta basada en ITINN (A-III).
- DOR+FTC/TAF o DOR/3TC/TDF se considera una alternativa a las pautas preferentes en elTAR de inicio (C-I).
- Se recomienda esperar a conocer el resultado del estudio genotípico de resistencias si seva a empezar el TAR con una pauta basada en ITINN para los que existe mayor prevalencia deresistencias transmitidas (EFV, RPV) (A- II) o con TDF/3TC/DOR en un paciente con historia de uso de PrEP (A-III).
- En pacientes con CVP <100.000 cop/mL la combinación RPV/FTC/TAF se considera unaalternativa a las pautas preferentes en el TAR de inicio (C-I).
- RPV no debe utilizarse en pacientes con CVP >100.000 cop/mL (A-I).
3.2.4. INHIBIDORES DE LA PROTEASA POTENCIADOS
En el TAR de inicio sólo se pueden usar IP cuando van potenciados con RTV o COBI. En la actualidad existen cinco IP/p teóricamente disponibles, aunque en la práctica clínica, el único IP/p usado hoy en día en el TAR inicial es DRV/p, por lo que en este apartado sólo nos referiremos aeste fármaco.
Los IP son inductores e inhibidores del citocromo P450 y frecuentemente pueden originarinteracciones farmacológicas.
Los IP se caracterizan por una elevada barrera genética que dificulta la selección de MR aun en situaciones desfavorables como la baja adherencia. Por otra parte, son los FAR con peor perfilmetabólico, asociándose a hiperlipidemia, aunque en menor medida en el caso de ATV y DRV.
DRV/p puede utilizarse en el TAR de inicio en dosis QD, bien en forma de un comprimido de 800 mgpotenciado con 100 mg de RTV, coformulado con 150 mg de COBI (DRV/c) o en un comprimido único con FTC/TAF (DRV/c/FTC/TAF).
El estudio ARTEMIS53 comparó DRV/r (800/100 mg, QD) frente a LPV/r en 689 pacientes que recibieron además FTC/TDF coformulados. A las 48 semanas DRV/r resultó no inferior a LPV/r. Un menor porcentaje de pacientes tratados con DRV/r presentó diarrea de grado 2-4 y se observaronmenores elevaciones de colesterol y triglicéridos que en los tratados con LPV/r. A las 96 semanas,DRV/r resultó superior a LPV/r (en el análisis TLOVR, aunque no en el snapshot). Un 4% de los pacientes de la rama de DRV/r y un 9% de la rama de LPV/r abandonaron el tratamiento asignado.
DRV/r se comparó con RAL en el estudio ACTG 525726 sin objetivar diferencias en el porcentaje de FV. En el análisis snapshot y en el análisis conjunto de la respuesta virológica y tolerabilidad, DRV/rfue inferior a RAL.
DRV/r también se mostró inferior a DTG en el ensayo clínico FLAMINGO21, debidofundamentalmente a una mayor tasa de efectos adversos y discontinuaciones por causas norelacionadas con el fármaco.
DRV/c se ha comparado frente a ABC/3TC/DTG en el estudio SYMTRI54. Un total de 316 pacientes naïve fueron aleatorizados a DRV/c/FTC/TAF o DTG/ABC/3TC. La mayoría de los pacientes eranvarones, HSH, con una media de linfocitos CD4+ alrededor de 400 células/μL (el 11% y 14% tenían un recuento de CD4+ <200 células/μL) y una mediana de CVP alrededor de 60.000 cop/mL (el40% tenían >100.000 cop/mL, sin diferencias entre los dos grupos). Tras 48 semanas, DRV/c/ FTC/TAF tuvosimilares resultados de eficacia que DTG/ABC/3TC (79% vs 82% en el análisis ITT), pero no alcanzóla no inferioridad (IC 95%: -11,3 a 6,6, p=0,706).
En el ensayo clínico aleatorizado y doble ciego AMBER se comparó DRV/c administrado junto conFTC/TDF o coformulado con FTC/TAF en comprimido único, en pacientes sin tratamiento previo25. La combinación DRV/c/FTC/TAF demostró una eficacia no inferior a las 48 semanas, aunque el estudio incluyó muy pocos pacientes con infección en fase avanzada (sólo 6% con linfocitos CD4+ <200 células/ μL). La tolerancia de ambas pautas fue buena, con pocas discontinuaciones por efectos adversos (4,4% y 2,2% respectivamente). La evolución de los marcadores de toxicidad renal, ósea yniveles de lípidos fue consistente con lo ya descrito cuando se utiliza TAF y/o COBI.
Debido al mayor riesgo de interacciones farmacológicas y no haber demostrado la no inferioridadrespecto a las combinaciones de TAR consideradas preferentes, las combinaciones con DRV/p sólo se recomiendan como alternativas en pautas de inicio.
Recomendaciones
- Actualmente no se considera preferente para TAR de inicio ninguna pauta basada en IP/p (A-III).
- Cuando se considere conveniente iniciar un tratamiento basado en IP se recomiendautilizar DRV/c/FTC/TAF (A-I) o DRV/r+FTC/TAF (QD) (A-III).
Tablas:

1Se consideran como excepción los pacientes que mantienen carga viral indetectable de forma mantenida sin TAR (controladores de élite). En este caso no existe información que permita valorar el efecto beneficioso del TAR, por lo que no se puede establecer una recomendación de tratamiento.
2La disposición y la motivación del paciente es un factor crítico a la hora de tomar la decisión de Es importante hacer una valoración individualizada del momento de inicio del TAR y de los FAR que deben formar parte del régimen inicial, sopesando las ventajas e inconvenientes de cada una de las opciones.
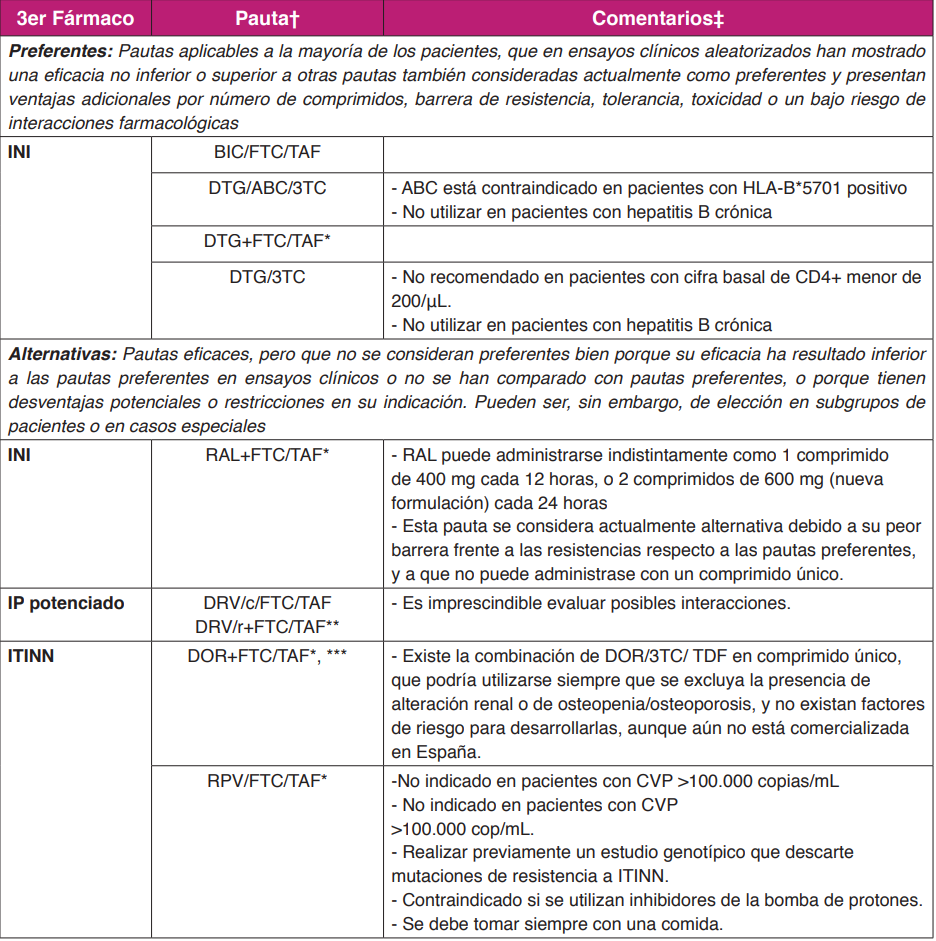
Notas:
† En el caso de personas embarazadas o de pacientes con tuberculosis estas recomendaciones no son válidas y se debe utilizar la información existente en los apartados correspondientes y las guías específicas.
† Si se opta por un inicio rápido tras el diagnóstico, es habitual no disponer del resultado del estudio de resistencias ni de la determinación de HLA-B*5701, por lo que no se deben utilizar regímenes basados en ITINN ni con ABC. Si se inicia el TAR antes de disponer de los resultados del recuento de linfocitos CD4+ o CVP hay que evitar de inicio los regímenes cuya recomendación esté condicionada por estos resultados (como los basados en RPV o la combinación DTG+3TC).
† Cuando estén disponibles, se recomienda el uso de preparados que combinen fármacos a dosis fijas. Los ensayos clínicos en los que se fundamenta la evidencia de cada pauta se referencian en el texto.
† Los comentarios reflejan aspectos que se deben considerar en la elección de régimen, pero no pretenden ser una guía exhaustiva de las precauciones a tomar en el uso de los fármacos. Para mayor información se recomienda revisar el texto del documento, así como las fichas técnicas de los fármacos.
† En otro apartado de estas guías se tratan aspectos de precio y de costes de los diferentes regímenesterapéuticos.
* La utilización de TFV como TDx puede considerarse una alternativa a TAF cuando no se asocie a un fármaco potenciado y siempre que se excluya la presencia de alteración renal o de osteopenia/osteoporosis, y no existan otros factores de riesgo para desarrollarlas.
** DRV se puede potenciar con RTV o COBI. La combinación con COBI disminuye el número de pastillas al estar disponible la combinación coformulada en un solo comprimido. En la elección de potenciador se deben revisar las posibles interacciones con RTV o COBI, que en ocasiones no coinciden.
*** DOR, y la combinación coformulada de DOR/3TC/TDF han sido aprobadas por la EMA. En España, sólo está aprobada DOR por separado.
Bibliografía:
Lundgren JD, Babiker AG, Gordin F, et al. Initiation of antiretroviral therapy in early asymptomatic HIV N Engl J Med 2015; 373:795-807.
Danel C, Moh R, Gabillard D, et al. A trial of early antiretrovirals and isoniazid preventive therapy in N Engl J Med 2015; 373:808-22.
Cohen MS, Chen YQ, McCauley M, et al. Antiretroviral therapy for the prevention of HIV-1 N Engl J Med 2016; 375:830-9.
Ford N, Migone C, Calmy A, et al. Benefits and risks of rapid initiation of antiretroviral AIDS2018; 32:17-23.
Girometti N, Nwokolo N, McOwan A, Whitlock G. Outcomes of acutely HIV-1- infected individualsfollowing rapid antiretroviral therapy Antivir Ther 2017; 22: 77-80.
Hoenigl M, Chaillon A, Moore DJ, et al. Rapid HIV viral load suppression in those initiating antiretroviraltherapy at first visit after HIV Sci Rep 2016; 6: 32947.
Pilcher CD, Ospina-Norvell C, Dasgupta A, Jones D, Hartogensis W, Torres S, et al. The effect of same-day observed initiation of antiretroviral therapy on HIV viral load and treatment outcomes in a US Public Health Setting. J Acquir Immune Defic Syndr 2017; 74:44-51.
Huhn GD, Crofoot G, Ramgopal M, Gathe J Jr, Bolan R, Luoet D, et al. Darunavir/Cobicistat/Emtricitabine/Tenofovir Alafenamide in a rapid-initiation model of care for humanimmunodeficiency virus type 1 infection: primary analysis of the DIAMOND Clin Infect Dis 2020;71:3110-7.
Rolle Ch-P, Berhe M, Singh T, et Dolutegravir/lamivudine as a first-line regimen in a test-and-treatsetting for newly diagnosed people living with HIV. AIDS 2021; 35:1957-65.
Rolle Ch-P, Berhe M, Singh T, et al. Feasibility, efficacy and safety of Dolutegravir/ lamivudine (DTG/3TC) as a first-line regimen in a test-and-treat setting for newly diagnosed people living with HIV (PLWH): 48-week results of STAT study. 11th IAS Conference on HIV Science; July 18-21, 2021;
Bachelard A, Isernia V, Charpentier C, et al. Efficacy and safety using bictegravir/emtricitabine/tenofovir alafenamide (BIC/FTC/TAF) in a test and treat model: the FAST 18thEuropean AIDS Conference. October 27th-30th, 2021. Online & London, UK.
Arribas JR, Thompson M, Sax PE, et al. Randomized, double-blind comparison of tenofovir alafenamide (TAF) vs tenofovir disoproxil fumarate (TDF), each coformulated with elvitegravir, cobicistat,and emtricitabine (E/C/F) for initial HIV-1 treatment: week 144 J Acquir Immune Defic Syndr 2017;75:211-8.
Venter WDF, Moorhouse M, Sokhela S, et Dolutegravir plus Two Different Prodrugs of Tenofovir toTreat HIV. N Engl J Med 2019; 381:803-15.
Hill A, Hughes SL, Gotham D, Pozniak AL. Tenofovir alafenamide versus tenofovir disoproxil fumarate: is there a true difference in efficacy and safety? J Virus Erad 2018; 4:72-79.
Gallant J, Lazzarin A, Mills A, et al. Bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir, abacavir, and lamivudine for initial treatment of HIV-1 infection (GS- US-380-1489): a double-blind, multicentre, phase 3, randomised controlled non-inferiority Lancet 2017; 390:2063-72.
Sax PE, Tierney C, Collier AC, et al. Abacavir-lamivudine versus tenofovir-emtricitabine for initial HIV-1 N Engl J Med 2009; 361:2230-40.
Sax PE, Tierney C, Collier AC, et Abacavir/lamivudine versus tenofovir DF/ emtricitabine as part of combination regimens for initial treatment of HIV: final results. J Infect Dis 2011; 204:1191-201.
Daar ES, Tierney C, Fischl MA, et al. Atazanavir plus ritonavir or efavirenz as part of a 3-drug regimen for initial treatment of HIV-1. Ann Intern Med 2011; 154:445-56.
Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment ofHIV-1 N Engl J Med 2013; 369:1807-18.
Raffi F, Rachlis A, Stellbrink HJ, et al. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, non-inferiority SPRING-2 study. Lancet 2013; 381:735-43.
Clotet B, Feinberg J, van Lunzen J, et Once-daily dolutegravir versus darunavir plus ritonavir in antiretroviral-naive adults with HIV-1 infection (FLAMINGO): 48 week results from the randomised open-label phase 3b study. Lancet 2014; 383:2222-31.
Cahn P, Kaplan R, Sax PE, et al. Raltegravir 1200 mg once daily versus raltegravir 400 mg twice daily, with tenofovir disoproxil fumarate and emtricitabine, for previously untreated HIV-1 infection: a randomised, double-blind, parallel-group, phase 3, non- inferiority The lancet HIV 2017; 4:e486-e94.
Lennox JL, Landovitz RJ, Ribaudo HJ, et al. Efficacy and tolerability of 3 nonnucleoside reversetranscriptase inhibitor-sparing antiretroviral regimens for treatment-naive volunteers infected with HIV- 1: A randomized, controlled equivalence trial. Ann Intern Med 2014; 161:461-71.
Rockstroh JK, DeJesus E, Lennox JL, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naive HIV-1- infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr 2013; 63:77-85.
Eron JJ, Orkin C, Gallant J, Molina JM, Negredo E, Antinori A, et al. A week-48 ran- domized phase-3trial of darunavir/cobicistat/ emtricitabine/tenofovir alafenamide in treatment-naive HIV-1 AIDS2018; 32:1431-42.
Walmsley S, Baumgarten A, Berenguer J, et al. Dolutegravir plus abacavir/lamivudine for the treatment of HIV-1 infection in antiretroviral therapy-naive patients: week 96 and week 144 results from the SINGLE randomized clinical trial. J Acquir Immune Defic Syndr 2015; 70:515-9.
NAMSAL ANRS 12313 Study Group, Kouanfack C, Mpoudi-Etame M, Omgba Bassega P, et Dolutegravir-based or low-dose efavirenz-based regimen for the treatment of HIV- 1. N Engl J Med 2019;381:816-26.
Molina JM, Clotet B, van Lunzen J, et al. Once-daily dolutegravir versus darunavir plus ritonavir fortreatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomised, open- label, phase 3b study. Lancet HIV 2015; 2:e127-36.
Orrell C, Hagins DP, Belonosova E, et al. Fixed-dose combination dolutegravir, abacavir, andlamivudine versus ritonavir-boosted atazanavir plus tenofovir disoproxil fumarate and emtricitabine in previously untreated women with HIV-1 infection (ARIA): week 48 results from a randomised, open-label, non- inferiority, phase 3b study. The Lancet HIV 2017; 4:e536-e546.
Cahn P, Sierra Madero J, Arribas JR, et al. Dolutegravir plus lamivudine versus dolutegravir plus tenofovir disoproxil fumarate and emtricitabine in antiretroviral-naive adults with HIV-1 infection (GEMINI-1and GEMINI-2): week 48 results from two multicentre, double-blind, randomised, non-inferiority, phase 3 Lancet 2019; 393:143-55.
Cahn P, Sierra Madero J, Arribas J, et al. Durable efficacy of dolutegravir (DTG) plus lamivudine (3TC) in antiretroviral treatment-naive adults with HIV-1 infection--96-week results from the GEMINI studies. 10th IAS Conference on HIV Science (IAS 2019), July 21-24, 2019, Mexico Abstract WEAB0404LB.
Orkin C, Porteiro N, Berhe M, et Durable efficacy of DTG+3TC in GEMINI-1&2: year 3 subgroup analysis. Conference on Retroviruses and Opportunistic Infections, March, 6-10, 2021; Virtual.
Radford M, Parks DC, Ferrante S, Punekar Y. Comparative efficacy and safety and dolutegravir andlamivudine in treatment naive HIV AIDS 2019; 33:1739-49.
Sax PE, Pozniak A, Montes ML, et al. Coformulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection (GS-US-380-1490): a randomised, double-blind, multicentre, phase 3, non-inferiority Lancet2017; 390:20.
Workowski K. Four-year outcomes of B/F/TAF in treatment-naïve adults. Conference on Retrovirusesand Opportunistic Infections, March 6-10, 2021;
Mollan KR, Smurzynski M, Eron JJ, et al. Association between efavirenz as initial therapy for HIV-1infection and increased risk for suicidal ideation or attempted or completed suicide: An analysis of trial Ann Intern Med 2014; 161:1-10.
Arenas-Pinto A, Grund B, Sharma S, et al. Risk of Suicidal Behavior With Use of Efavirenz: Results from the Strategic Timing of Antiretroviral Treatment Trial. Clin Infect Dis 2018; 67:420-9.
Smith C, Ryom L, Monforte A, et Lack of association between use of efavirenz and death fromsuicide: evidence from the D:A:D study. J Int AIDS Soc 2014; 17:19512.
Napoli AA, Wood JJ, Coumbis JJ, Soitkar AM, Seekins DW, Tilson HH. No evident association between efavirenz use and suicidality was identified from a disproportionality analysis using the FAERS J Int AIDS Soc 2014; 17:19214.
Van Leth F, Phanuphak P, Stroes E, et al. Nevirapine and efavirenz elicit different changes in lipid profiles in antiretroviral-therapy-naive patients infected with HIV-1. PLoS Medicine 2004; 1:e19.
Riddler SA, Haubrich R, DiRienzo AG, et Class-sparing regimens for initial treatment of HIV-1infection. N Engl J Med 2008; 358:2095-106.
Montaner JS, Schutz M, Schwartz R, et Efficacy, safety and pharmacokinetics of once-daily saquinavir soft-gelatin capsule/ritonavir in antiretroviral-naive, HIV-infected patients. MedGenMed:Medscape general medicine 2006;8:36.
Bartlett JA, Johnson J, Herrera G, et al. Long-term results of initial therapy with abacavir and lamivudine combined with efavirenz, amprenavir/ritonavir, or stavudine. J Acquir Immune Defic Syndr2006; 43:284-92.
Smith KY, Tierney C, Mollan K, et al. Outcomes by sex following treatment initiation with atazanavir plus ritonavir or efavirenz with abacavir/lamivudine or tenofovir/emtricitabine. Clin Infect Dis 2014; 58:555-63.
Molina JM, Cahn P, Grinsztejn B, et al. Rilpivirine versus efavirenz with tenofovir and emtricitabine intreatment-naive adults infected with HIV-1 (ECHO): a phase 3 randomised double-blind active-controlled Lancet 2011; 378:238-46.
Cohen CJ, Andrade-Villanueva J, Clotet B, et al. Rilpivirine versus efavirenz with two background nucleoside or nucleotide reverse transcriptase inhibitors in treatment-naive adults infected with HIV-1 (THRIVE): a phase 3, randomised, non-inferiority trial. Lancet 2011; 378:229-37.
Nelson MR, Elion RA, Cohen CJ, et al. Rilpivirine versus efavirenz in HIV-1-infected subjects receivingemtricitabine/tenofovir DF: pooled 96-week data from ECHO and THRIVE HIV Clin Trials 2013;14:81-91.
Cohen C, Wohl D, Arribas JR, et al. Week 48 results from a randomized clinical trial of rilpivirine/ emtricitabine/tenofovir disoproxil fumarate vs. efavirenz/emtricitabine/tenofovir disoproxil fumarate intreatment-naive HIV-1-infected AIDS 2014; 28:989-97.
Hagins D, Orkin C, Daar ES, et Switching to coformulated rilpivirine (RPV), emtricitabine (FTC) and tenofovir alafenamide from either RPV,FTC and tenofovir disoproxil fumarate (TDF) orefavirenz, FTC and TDF: 96-week results from two randomized clinical trials. HIV Med. 2018; 19:724-33.
Orkin C, Squires KE, Molina JM, et al. Doravirine/Lamivudine/Tenofovir Disoproxil Fuma-rate is Non- inferior to Efavirenz/Emtricitabine/ Tenofovir Disoproxil Fumarate in Treatment-naive Adults With HumanImmunodeficiency Virus-1 Infection: Week 48 Results of the DRIVE-AHEAD Clin Infect Dis 2019;68:535-44.
Molina JM, Squires K, Sax PE, et Doravirine versus ritonavir-boosted darunavir in antiretroviral-naive adults with HIV-1 (DRIVE-FORWARD): 48-week results of a randomised, double-blind, phase 3, non-inferiority trial. The Lancet HIV 2018; 5:e211-e220.
Molina JM, Squires K, Sax PE, et Doravirine versus ritonavir-boosted darunavir: 96- week results ofthe randomised, double-blind, phase 3 DRIVE-FORWARD, non-inferiority trial. In: International AIDSConference (AIDS2018), Amsterdam, The Netherlands, 23-27 July 2018.
Orkin C, DeJesus E, Khanlou H, et Final 192-week efficacy and safety of once-daily darunavir/ ritonavir compared with lopinavir/ritonavir in HIV-1-infected treatment-naive patients in the ARTEMIS trial.HIV Med 2013; 14:49-59.
Podzamczer D, Micán R, Tiraboschi J, et Darunavir/cobicistat/emtricitabine/ tenofovir alafenamideversusdolutegravir/abacavir/lamivudine in antiretroviral-naïve adults (SYMTRI): a multicenter randomized opten-label study (PreEC/RIS-57). Opten Fórum Infec Dis (en prensa).
4. CAMBIO DEL TAR EN PACIENES CON REPLICACIÓN VIRAL SUPRIMIDA
4.1. CAMBIO DE FÁRMACOS ANTIRRETROVIRALES. CONSIDERACIONES
4.2. CAMBIOS A REGÍMENES QUE SIGUEN INCLUYENDO TRES FÁRMACOS ANTIRRETROVIRALES (TABLA 5)
4.3. CAMBIOS A REGÍMENES QUE INCLUYEN MENOS DE TRES FÁRMACOS ANTIRRETROVIRALES (TABLA 5).
4.4. SEGUIMIENTO POSTERIOR A UN CAMBIO DE TAR EN PACIENTES CON REPLICACIÓN VIRAL SUPRIMIDA
En este capítulo se revisan las opciones de cambio del TAR en pacientes que tienen la CVP indetectable.
Motivos para cambiar un TAR eficaz
Existen muchos motivos para cambiar un TAR eficaz: toxicidad, nuevas comorbilidades, interacciones farmacológicas, mejora de la posología, de dosis diarias o del número de
medicamentos, preferencias de los pacientes, requerimientos dietéticos, embarazo y coste del propio TAR. El cambio puede ser proactivo cuando se realiza preventivamente o reactivo cuando el régimen actual ha dejado de ser el ideal para el paciente debido a alguno de los motivos reseñados.
Definición de carga viral suprimida
La mayoría de los ensayos clínicos de cambio de TAR han incluido pacientes con CVP menor de 50 cop/mL durante al menos 6 meses, por lo que sus resultados son aplicables preferiblemente a pacientes con un tiempo de supresión similar. Como norma general, cuanto más prolongado sea el período de supresión virológica es menos probable que el cambio de TAR se asocie a FV.
Objetivo del cambio del TAR eficaz
El objetivo es mantener la supresión virológica y optimizar el TAR de acuerdo a las características y la preferencia del paciente.
Circunstancias que obligan a cambiar el TAR eficaz
El cambio proactivo es obligado cuando evidencias sólidas avalan que el paciente tiene más riesgo de presentar un efecto adverso grave o irrecuperable si se mantiene el TAR actual que si se cambia. Un ejemplo sería la presencia de interacciones farmacológicas significativas consecuencia del inicio de tratamiento con rifampicina en pacientes que toman IP/p.
El cambio reactivo es obligado si el efecto adverso va a desaparecer tras el cambio de TAR, como por ejemplo los efectos adversos del SNC causados por EFV.
Es obvio que si un paciente tiene la carga viral suprimida es porque es capaz de continuar tomando la pauta prescrita. El clínico no debe olvidar que en ocasiones ese nivel de adherencia se consigue gracias a un sobresfuerzo del paciente, que es capaz de sobrellevar efectos adversos que pueden ser erróneamente entendidos como inevitables.
Recomendación:
- No se debe asumir que un TAR es óptimo sólo porque mantenga la carga viral suprimida. En todas las revisiones se debe preguntar con detalle sobre el esfuerzo que necesita hacer el paciente para adherirse al TAR pautado (A-III).
CAMBIO DE FÁRMACOS ANTIRRETROVIRALES. CONSIDERACIONES
Consideraciones sobre el nuevo régimen
Recomendaciones
- En pacientes con CVP suprimida el nuevo régimen debe priorizar combinaciones que han demostrado beneficio en ensayos clínicos diseñados específicamente para valorar el cambio en estos pacientes o en ensayos clínicos en pacientes naive, idealmente con fármacos de los recomendados como preferentes en pacientes sin TAR previo (A-I).
- En casos seleccionados los regímenes alternativos en pacientes sin TAR previo (Tabla 3) pueden ser apropiados (A-III).
Consideraciones virológicas
Tras el cambio del TAR el mantenimiento de la supresión virológica es la norma en los pacientes sin historia de FV, aún cuando se cambie de una pauta de alta barrera genética a una pauta de baja barrera genética. Cambiar el TAR es más complicado en pacientes con FV previos que pueden haber causado y archivado MR, especialmente si se trata de cambiar un TAR que incluye IP/p o un INI de alta barrera genética (DTG y BIC).
Datos preliminares sugieren que pudiera no ser necesaria la plena actividad de alguno de los dos ITIAN cuando se cambia a pautas que contienen DTG o BIC, especialmente si sólo está presente la mutación M184V/I. Aún son necesarios más datos para conocer el impacto real en la eficacia de estos cambios a largo plazo.1234
Recomendaciones
- Antes de cambiar un TAR eficaz debe realizarse una evaluación minuciosa del perfil de fracasos previos y resistencias del paciente, así como de toxicidades, interacciones, restricciones dietéticas y actividad sobre el VHB, si fuera necesario, del nuevo régimen (AIII).
- El cambio desde una pauta doble o triple basada en un IP/r o un INI de alta barrera genética a otra basada en un ITINN o INI de baja barrera genética solo debe hacerse si se puede garantizar la actividad antiviral de todos los fármacos del nuevo régimen (A-I).
- Si se piensa suspender TFV (TDF ó TAF) deberá revisarse previamente que no existe infección activa por el VHB (A-III). En caso de hepatitis B crónica, si la suspensión está indicada, valorar añadir un fármaco activo (como entecavir) a la nueva pauta de TAR.
- En el caso de los pacientes sin inmunidad o que no han respondido a la vacunación frente al VHB y con alto riesgo de infección, hay que priorizar la actividad frente a VHB del nuevo régimen y valorar de forma individual la indicación de pautas duales sin actividad frente al VHB (A-III).
Consideraciones sobre la gradación de la evidencia
Se considera que existen datos suficientes para hacer una recomendación fuerte de cambio de antirretrovirales en los escenarios contemplados en la Tabla 2.
Este panel distingue entre la fuerza de la recomendación para cambiar el TAR o para priorizar una pauta alternativa. Recomendamos que el clínico consulte la (Tabla 4) para intentar responder a la pregunta ¿Debo cambiar el TAR?, y una vez establecida la necesidad de cambio, la (Tabla 5) gradúa la evidencia para recomendar una nueva pauta.
La recomendación para avalar el cambio de TAR puede ser débil pero una vez que el cambio se ha decidido la recomendación sobre la pauta a la que cambiar puede ser fuerte. Un ejemplo apropiado es el cambio desde múltiples comprimidos a una pastilla única simplemente con el objetivo de simplificación. Aunque hay evidencia preliminar de que el TAR con pastilla única podría aumentar la adherencia y disminuir las hospitalizaciones5, en este momento el comité no considera que exista evidencia definitiva para hacer una recomendación fuerte que respalde este cambio en la mayoría de los pacientes. Sin embargo, si el clínico ha decidido que en un paciente la simplificación posológica está indicada, entonces el comité hace una recomendación fuerte sobre las pautas más apropiadas.
CAMBIOS A REGÍMENES QUE SIGUEN INCLUYENDO TRES FÁRMACOS ANTIRRETROVIRALES (TABLA 5)
4.2.1 CAMBIO DE ITIAN
Cambio de ABC/3TC a TDF/FTC o TAF/FTC
Fundamento. Un ensayo clínico aleatorizado6 ha demostrado que el cambio de ABC/3TC a TDF/FTC es seguro virológicamente y produce una mejoría del perfil lipídico. El cambio a TDF/FTC se asocia a una disminución del FGe, especialmente si el tercer fármaco es un IP/p.
Un ensayo clínico aleatorizado7 ha demostrado que el cambio de ABC/3TC a TAF/ FTC es seguro virológicamente pero no se asocia a beneficios en perfil lipídico, función renal o densidad mineral ósea.
La relación de ABC con un incremento en la incidencia de eventos cardiovasculares es muy controvertida.
Recomendación
- El cambio de ABC/3TC a TDF/FTC o a TAF/FTC puede realizarse por decisión clínica, pero el cambio no se puede recomendar con el objetivo de reducir el riesgo cardiovascular (C-I).
Cambio de TDF a ABC
Fundamento. TDF se ha asociado con una disminución de la densidad mineral ósea. En un pequeño ensayo clínico aleatorizado en pacientes con osteopenia u osteoporosis, el cambio de TDF a ABC se siguió de un incremento de la densidad mineral ósea en el fémur8.
Recomendación
- En pacientes con osteopenia u osteoporosis asociada al uso de TDF, este fármaco debe suspenderse. El cambio por ABC es adecuado (A-II).
Cambio de TDF a TAF
Fundamento. Dos ensayos clínicos aleatorizados y doble ciego muestran que el cambio de TDF por TAF es seguro virológicamente, demostrándose no inferioridad en cuanto a eficacia frente a continuar con el tratamiento previo, y se asocia a un incremento significativo de la densidad mineral ósea en el fémur y columna vertebral, así como de una mejoría en parámetros de función renal y tubular y a un ligero empeoramiento del perfil lipídico910. En el primero se incluyen pacientes tratados con TDF/FTC y un tercer farmaco y se cambia TDF/FTC por TAF/FTC9. En el segundo se incluyen pacientes en tratamiento con RPV/FTC/TDF y se cambia por RPV/FTC/TAF10. El cambio a TAF/ FTC es eficaz para mantener la supresión del VHB en pacientes coinfectados11.
Recomendación
- En pacientes tratados con pautas basadas en TDF y con alteraciones significativas de la función renal o de la densidad mineral ósea u otros factores de riesgo para desarrollarlas, este fármaco debe suspenderse. El cambio a una pauta basada en TAF es una opción adecuada (A-I).
4.2.2. CAMBIO A REGÍMENES BASADOS EN ITINN
Cambio de EFV a RPV o DOR
Fundamento. Dos ensayos clínicos aleatorizados han demostrado que el cambio desde EFV/FTC/TDF a RPV/FTC/TDF12 o a RPV/FTC/TAF13 es no inferior frente a continuar el tratamiento previo en cuanto a eficacia. Cuando sólo se cambió EFV por RPV mejoró el perfil lipídico y la toxicidad persistente del SNC. Además, el cambio de TDF por TAF se asoció a un incremento de la densidad mineral ósea en el fémur y la columna vertebral, así como a una mejoría en parámetros de función renal y tubular.
Un ensayo clínico aleatorizado y abierto14 ha demostrado que el cambio desde diferentes pautas con EFV a DOR/3TC/TDF es no inferior a continuar con el tratamiento previo. En otro ensayo clínico, aleatorizado y doble ciego, realizado en pacientes con control virológico al menos durante 12 semanas, que recibían EFV/FTC/TDF y presentaban toxicidad del SNC de grado 2 o mayor atribuible a EFV, el cambio a la combinación de DOR/3TC/TDF no produjo mejoría significativa comparado con seguir con EFV/FTC/TDF. La eficacia virológica se mantuvo a las 24 semanas y el perfil lipídico mejoró14.
No hay datos para recomendar un cambio proactivo de EFV para evitar la toxicidad del SNC en pacientes sin síntomas del SNC.
Recomendación
- En pacientes con efectos adversos del SNC causados por EFV, éste fármaco debe suspenderse. El cambio a RPV es una opción adecuada (A-I).
- En pacientes con dislipemia que estén tomando pautas que incluyan EFV, el cambio de la pauta a DOR/FTC/TDF es adecuado (A-I)
Cambio de IP/p a RPV o a DOR
Fundamento. Un ensayo clínico abierto ha demostrado que el cambio desde dos ITIAN más un IP/p a RPV/FTC/TDF es seguro virológicamente y se asocia a una mejoría en el perfil lipídico (CT, C-LDL, cociente CT/C-HDL y TG) y a una mejoría en los efectos adversos gastrointestinales causados por los IP/p16.
No hay ningún ensayo clínico que analice directamente el cambio desde dos ITIAN más un IP/p a RPV/FTC/TAF, pero sí evidencia indirecta, dada la eficacia del cambio desde dos ITIAN más un IP a RPV/FTC/TDF16 y de RPV/FTC/TDF a RPV/FTC/ TAF12.
Un ensayo clínico aleatorizado14 ha demostrado que en pacientes con supresión virológica el cambio a DOR/3TC/TDF desde 2 ITIAN más un IP/p, un ITINN o un INI (muy pocos con INI previo) es no inferior a continuar con el tratamiento previo. En el subgrupo de pacientes con IP previo, el cambio se asoció a beneficios en el perfil lipídico.
Recomendación
- En pacientes con alteraciones gastrointestinales asociadas a IP/p estos fármacos deben suspenderse. El cambio de la pauta a RPV/FTC/TDF (A-I), a RPV/FTC/TAF (A-III) o a DOR/FTC/TDF (A-I) es adecuado.
- En pacientes con dislipemia asociada a IP/p la suspensión de estos fármacos es una opción terapéutica. El cambio de la pauta a RPV/FTC/TDF (A-I), a RPV/ FTC/TAF (A-III) o a DOR/FTC/TDF (A-I) es adecuado.
4.2.3 CAMBIO A REGÍMENES BASADOS EN IP/P
En este escenario, disponemos de evidencia científica del cambio a partir de pautas que ya contienen un IP/p.
Cambio de IP/p a DRV/c
Fundamento. Este cambio de potenciador está basado en los resultados de DRV/c en un ensayo clínico de brazo único17.
Los resultados de los ensayos de los estudios de bioequivalencia hacen que este comité recomiende el uso de DRV/c en los escenarios descritos en este capítulo que afectan a DRV/r como parte de regímenes triples. En regímenes dobles no hay ensayos clínicos del cambio de DRV/r a DRV/c pero algunos estudios de cohorte han mostrado que el cambio de DRV/r a DRV/c es seguro y se mantiene la eficacia en el control de la replicación viral1819.
Deben revisarse en todo caso las posibles interacciones con otros fármacos puesto que no son idénticas con RTV y con COBI.
Un ensayo clínico aleatorizado20 ha demostrado que en pacientes sin fracasos previos a DRV y sin historia de resistencia a DRV, aunque con posibles fallos virológicos a otras pautas, el cambio de IP/p+TDF/FTC a DRV/c/FTC/TAF es virológicamente seguro y se asocia a disminución de proteinuria y mejoría de la densidad mineral ósea.
Recomendación
- En pacientes tratados con IP/p con riesgo de adherencia selectiva o deseo de reducir el número de comprimidos se debe cambiar la pauta. El cambio de DRV/r a DRV/c y de cualquier IP/p + 2 ITIAN a DRV/c/FTC/TAF son opciones adecuadas (A-I).
4.2.4 CAMBIO A REGÍMENES BASADOS EN INI
4.2.4.1 CAMBIO A REGÍMENES BASADOS EN RALTEGRAVIR
Cambio de EFV a RAL
Fundamento. En pacientes que toleran EFV, un ensayo clínico aleatorizado y doble ciego ha demostrado que el cambio de EFV a RAL mejora los niveles de lípidos (CT, C-LDL, TG) y en algunos pacientes mejora los scores de ansiedad y de estrés, manteniendo la supresión virológica21. No hay datos para recomendar un cambio proactivo de EFV para evitar la toxicidad del SNC en pacientes sin síntomas del SNC. No hay datos comparativos del cambio de EFV por RAL frente al cambio por otros antirretrovirales que tampoco causan síntomas del SNC.
Recomendaciones
- En pacientes con efectos adversos del SNC causados por EFV, éste fármaco debe suspenderse. El cambio a RAL es una opción adecuada (A-I).
- En pacientes con dislipemia asociada a EFV la suspensión de este fármaco es una opción terapéutica. El cambio a RAL es adecuado (A-I).
Cambio de IP/p a RAL
Fundamento. Tres ensayos clínicos aleatorizados han demostrado que el cambio de IP/p a RAL es seguro virológicamente si los dos ITIAN son completamente activos. El cambio se asocia en una mejoría de los niveles de CT, C-LDL, cociente CT/C-HDL y TG2223. Los resultados del estudio SPIRAL23 sugieren que si el tiempo de supresión viral es muy prolongado, el riesgo de fracaso virológico es menor, independientemente de la actividad de los ITIAN.
Recomendación
En pacientes tratados con IP/p y dislipemia la suspensión del IP/p es una opción terapéutica. El cambio a una pauta basada en RAL es una opción adecuada (A- I).
4.2.4.2 CAMBIO A REGÍMENES BASADOS EN DOLUTEGRAVIR
Cambio a DTG/ABC/3TC desde pautas que contienen IP, ITINN o INI + 2 ITIAN
Fundamento. Un ensayo clínico abierto ha demostrado que en pacientes con replicación viral suprimida que están recibiendo IP, ITNN o INI + 2 ITIAN, el cambio a DTG/ABC/3TC es virológicamente no-inferior24. El estudio demostró una mejoría en las escalas subjetivas de satisfacción con el tratamiento, pero no en otras variables clínicas.
Recomendación
- En pacientes tratados con IP/p, ITINN o INI + 2 ITIAN que quieran simplificar su régimen el cambio a una pauta de comprimido único es una opción terapéutica. El cambio a DTG/ABC/3TC es una opción adecuada si el HLA-B*5701 es negativo (A-I).
Cambio de IP/p+2 ITIAN a DTG+2 ITIAN
Fundamento. Un ensayo clínico aleatorizado y abierto con pacientes con replicación viral suprimida, sin historia de FV, mayores de 50 años y/o un score de Framingham >10% ha demostrado que el cambio de 2 ITIAN más un IP/p (fundamentalmente ATV/r, DRV/r) a DTG+2 ITIAN es seguro virológicamente. Los pacientes que cambiaron de tratamiento mejoraron significativamente el perfil lipídico25.
Recomendación
- En pacientes tratados con IP/p y dislipemia la suspensión del IP/p es una opción terapéutica. El cambio a una pauta basada en DTG es una opción adecuada (A-I).
4.2.4.3 CAMBIO A BIC/FTC/TAF
Cambio a BIC/FTC/TAF desde pautas que contienen un IP/p +2 ITIAN, DTG/ ABC/3TC o EVG/c/FTC/TDF
Fundamento. Tres ensayos clínicos aleatorizados262728 han demostrado que el cambio a BIC/FTC/TAF desde diferentes regímenes en pacientes con supresión virológica es no inferior a continuar con el tratamiento previo. En el primero26 se incluyeron pacientes en tratamiento con DRV/p o ATV/p + 2 ITIAN y se demuestra la no-inferioridad virológica y ligera mejoría del perfil lipídico y de la proteinuria tubular al cambiar a BIC/FTC/TAF. En el segundo27 se incluyeron pacientes en tratamiento con DTG/ABC/3TC y se demuestra la no inferioridad del cambio, pero no se asocia a beneficios en el perfil lipídico, función renal o densidad mineral ósea. En el tercero28 se incluyeron únicamente mujeres en tratamiento con EVG/c/FTC/TAF, EVG/c/FTC/TDF o ATV/r + TDF/FTC, demostrándose también la no-inferioridad del cambio.
El cambio a BIC/FTC/TAF en pacientes con supresión virológica se ha mostrado eficaz y no inferior a continuar con el tratamiento previo basado en IP/p o DTG, incluso en presencia de MR a ITIAN archivadas29.
Recomendación
- En pacientes tratados con IP/p + 2 ITIAN con alteraciones renales u óseas significativas asociadas a TDF o riesgo de desarrollarlas, TDF debe suspenderse. El cambio a BIC/FTC/TAF es una opción adecuada (A-I).
- En pacientes tratados con IP/p y dislipemia la suspensión del IP/p es una opción terapéutica. El cambio a BIC/FTC/TAF es una opción adecuada (A-I).
- El cambio de DTG/ABC/3TC a BIC/FTC/TAF puede realizarse por decisión clínica, pero no puede recomendarse el cambio buscando un beneficio en perfil lipídico, función renal o densidad mineral ósea, ni una reducción del riesgo cardiovascular (C-I).
CAMBIOS A REGÍMENES QUE INCLUYEN MENOS DE TRES FÁRMACOS ANTIRRETROVIRALES (TABLA 5).
En este escenario hay que tener en cuenta que ninguna de las pautas recomendadas es eficaz frente a la infección crónica por VHB y es necesario descartarla si se piensa suspender TFV (TDF o TAF).
4.3.1 TERAPIA DUAL CON IP/P + 3TC
Fundamento. Un meta-análisis de datos de pacientes individuales ha demostrado que la terapia dual con un IP/p+3TC es no inferior a la terapia triple con un IP/p+2 ITIAN y sin mayor riesgo de desarrollar resistencias30.
Recomendación
- En pacientes en tratamiento con IP/p + 2 ITIAN que quieran simplificar el TAR y/o evitar los efectos adversos causados por los ITIAN, el cambio a DRV/p + 3TC es una opción adecuada (A-I).
4.3.2 TERAPIA DUAL CON DTG + RPV
Cambio a DTG + RPV desde pautas que contienen IP/p, ITNN o INI + 2 ITIAN
Fundamento. Dos ensayos clínicos abiertos han demostrado que en pacientes con replicación viral suprimida durante al menos 12 meses y sin fracaso viral previo, que están recibiendo 2 ITIAN + IP/p, ITINN o INI, el cambio a DTG + RPV es virológicamente no-inferior a las 48 semanas31. A los 3 años de tratamiento se mantiene una elevada eficacia de la terapia dual y una baja tasa de fracaso virológico. Un 1.1% de los participantes tuvieron un fracaso virológico con selección de resistencias a rilpivirina32. Con DTG + RPV se demostró una mejoría en los biomarcadores de recambio óseo y en la densidad mineral ósea, no se observaron diferencias en los marcadores infamatorios, entre la terapia dual y el comparador, y hubo mas interrupciones debidas a efectos adversos con DTG + RPV (3% vs. <1%).
Recomendación
- En pacientes en tratamiento con IP/p, ITNN o INI + 2 ITIAN que quieran simplificar el TAR y/o evitar los efectos adversos causados por los ITIAN y nohayan presentado fracasos virológicos previos, el cambio a DTG + RPV es una opción adecuada (A-I).
4.3.3. TERAPIA DUAL CON DTG + 3TC
Cambio a DTG + 3TC desde pautas que contienen IP/p, ITINN o INI + 2 ITIAN
Fundamento. Dos ensayos clínicos aleatorizados fase 3, han demostrado que en pacientes con supresión virológica durante al menos 6 meses y sin fallos virológicos previos, el cambio a DTG+3TC es no inferior a continuar el TAR previo a las 48 semanas, sin que se desarrollaran MR en ningún paciente3334. El cambio a DTG+3TC se ha asociado a la mejoría de parámetros metabólicos (perfil lipídico y resistencia a la insulina) en aquellos pacientes que tomaban previamente un TAR con potenciadores35.
Recomendación
- En pacientes en tratamiento con IP/p, ITNN o INI + 2 ITIAN que quieran simplificar o evitar los efectos adversos causados por los ITIAN o los potenciadores del TAR, y sin resistencia conocida o sospechada a inhibidores de integrasa o a lamivudina, el cambio a DTG + 3TC es una opción adecuada (A-I).
4.3.4. TERAPIA DUAL CON DTG + DRV/P
Cambio a DTG + DRV/p desde diferentes pautas
Fundamento. Un estudio aleatorizado fase 3 ha mostrado que en pacientes en tratamiento con DRV/r + 2 ITIAN con supresión virológica durante al menos 6 meses, y con presencia de resistencias previas a ITIAN (9.1%), ITINN (12.9%) o IP (3.4%), el cambio a DTG+DRV/r es no inferior a continuar el TAR previo, sin que se desarrollen MR en ningún paciente3637.
Recomendación
- El cambio a DTG + DRV/p puede ser una opción en determinados pacientes que quieran simplificar su régimen actual (A-I) o evitar los efectos adversos asociados (A-II).
4.3.5. TERAPIA DUAL CON CAB+RPV AP
La administración del TAR en formulaciones de acción prolongada por vía I.M. ofrece beneficios potenciales para la calidad de vida de las personas con VIH o puede ser necesaria en alguna situación concreta. Algunas de las ventajas que ofrece son: menor frecuencia de dosis, eludir toxicidad gastrointestinal o interacciones asociadas al TAR, reducción del estigma o de la preocupación por la revelación del estatus VIH asociado a la toma de pastillas, el recuerdo diario de la condición de persona con VIH o evitar la preocupación de viajar a países con leyes restrictivas
AP hay que considerar varios factores:
- El uso de una fase inicial de tratamiento oral con cabotegravir y rilpivirina durante un mes no es imprescindible.
- Las poblaciones que han participado en los ensayos clínicos se caracterizaron por su buena adherencia y carga vírica indetectable mantenida, factores a tener en cuenta en la selección de candidatos.
- La frecuencia de resistencias a ITINN o INI en los fracasos virológicos, es mayor de la observada tradicionalmente con TAR oral basado en IP/p o INI de alta barrera genética.
- La larga vida de eliminación de CAB+RPV AP, podría favorecer la aparición de fracasos virológicos con resistencias en pacientes mal adherentes o que abandonen el TAR.
- El efecto adverso más común (>75%) con la combinación de CAB+RPV AP son las reacciones locales a las inyecciones, que se van tolerando mejor con las dosis sucesivas, y muy raramente (≤2%) causan la interrupción del tratamiento.
- Esta combinación no tiene efecto frente a VHB y no dispone de información sobre eficacia y seguridad en personas embarazadas.
- La administración parenteral de CAB+RPV AP no evita tener en cuenta las interacciones en el metabolismo con otros fármacos descritas con la administración oral de cualquiera de ellos, como los inductores de CYP3A. Por el contrario, soslaya tener en cuenta las interacciones descritas en la absorción gástrica, como el caso de inhibidores de la bomba de protones y rilpivirina.
Cambio a CAB+RPV AP desde pautas que contienen IP/p, ITINN o INI + 2 ITIAN
La administración de CAB+RPV AP como simplificación en pacientes con TAR estable, sin FV previos ni resistencias a INI o ITINN, sin infección activa por VHB y administración de CAB +RPV AP una vez al mes frente a continuar con el TAR por vía oral. Hubo 3 casos de fracaso virológico con CAB+RPV AP (3 con resistencias a RPV y uno además a INI) vs 4 casos con el TAR estable (3 de los cuales presentaron resistencias a inhibidores de la transcriptasa inversa). A las 96 semanas, ninguno de los 23 participantes que se mantuvieron en la pauta mensual de CAB +RPV AP entre dos pautas de administración I.M. de CAB+RPV AP, cada 4 y 8 semanas, demostrándose la no-inferioridad a las 48 semanas de la pauta administrada cada 8 semanas. Hubo 8 casos de fracaso virológico en el brazo de 8 semanas (6 con resistencias a ITINN, cinco de los cuales tenían también a INI) vs 2 casos en el de 4 semanas (ambos con MR a ITINN y INI). La no-inferioridad se mantuvo a las 96 semanas y sólo hubo un caso adicional de fracaso virológico (con MR a ITINN) que se dio en la pauta de 8 semanas42.
Además, en el ensayo clínico FLAIR43 se exploró el cambio precoz a una pauta I.M. en pacientes sin TAR previo que iniciaron DTG/ABC/3TC. A las 20 semanas, los pacientes con CV<50 cop/mL en semana 16, fueron aleatorizados a continuar con la pauta oral o cambiar a CAB+RPV AP i.m cada 4 semanas demostrándose la no- inferioridad a las 48 semanas. Tres de los cuatro pacientes con fracaso virológico en la pauta I.M. desarrollaron resistencias a ITINN e INI, por ninguno de los tres pacientes con fracaso virológico en la pauta oral. La eficacia y seguridad de la pauta de 4 semanas I.M. se mantuvo a las 96 semanas, detectándose un fracaso virológico adicional, en el brazo de DTG/ABC/3TC, que no presentó MR44.
En todos estos ensayos clínicos el efecto adverso más común fueron las reacciones en el lugar de inyección, las cuales disminuyeron en intensidad con el paso del tiempo y supusieron ≤2% de interrupciones del tratamiento.
Un análisis conjunto de los datos de estos tres estudios, que incluyó 1039 sujetos (13 con fracaso virológico), ha permitido identificar que la presencia de tres factores previos al inicio del TAR (resistencias a RPV archivadas en ADN proviral, los subtipos A6/A1 del VIH-1 y un IMC ≥30Kg/m2) y los niveles bajos de RPV en semana 8 se asocian a una mayor frecuencia de fracaso virológico. De los factores previos al inicio del TAR, la presencia de al menos dos de ellos se asocia a mayor riesgo de fracaso virológico (25.7%) comparado con pacientes sin ninguno de ellos (0.41%) o con 1 factor 0.37%. Sólo hubo 1 paciente con tres factores, y tuvo fracaso virológico.
Recomendación
- El cambio a CAB+RPV AP puede ser una opción para pacientes con TAR oral, sin fallo virológico ni evidencia de resistencia a ITINN o INI, y con carga viral indetectable durante un mínimo de 3-6 meses, que deseen o necesiten cambiar a una vía de administración alternativa. Este cambio implica un compromiso estrecho con el seguimiento clínico y la adherencia a las visitas de administración (A- I).
- Para evitar el riesgo de desarrollo de resistencias, si se interrumpe el TAR con CAB+RPV AP en un paciente con viremia suprimida es imprescindible iniciar una pauta de TAR supresora al mes o a los dos meses de la interrupción, según se administraran las inyecciones I.M. mensual o bimestralmente (AI). Si se interrumpe por sospecha o fracaso virológico confirmado debe instaurase un régimen alternativo eficaz sin demora (AIII).
SEGUIMIENTO POSTERIOR A UN CAMBIO DE TAR EN PACIENTES CON REPLICACIÓN VIRAL SUPRIMIDA
Recomendación
- Tras el cambio de un TAR es recomendable evaluar en un plazo de 3-6 semanas el mantenimiento de la supresión virológica y las determinaciones de laboratorio pertinentes, que dependerán del motivo del cambio (lipidograma, función renal, etc.). Una vez demostrada la continuación de la supresión virológica el paciente puede volver a la rutina de visitas habituales (B-III).
Tablas:
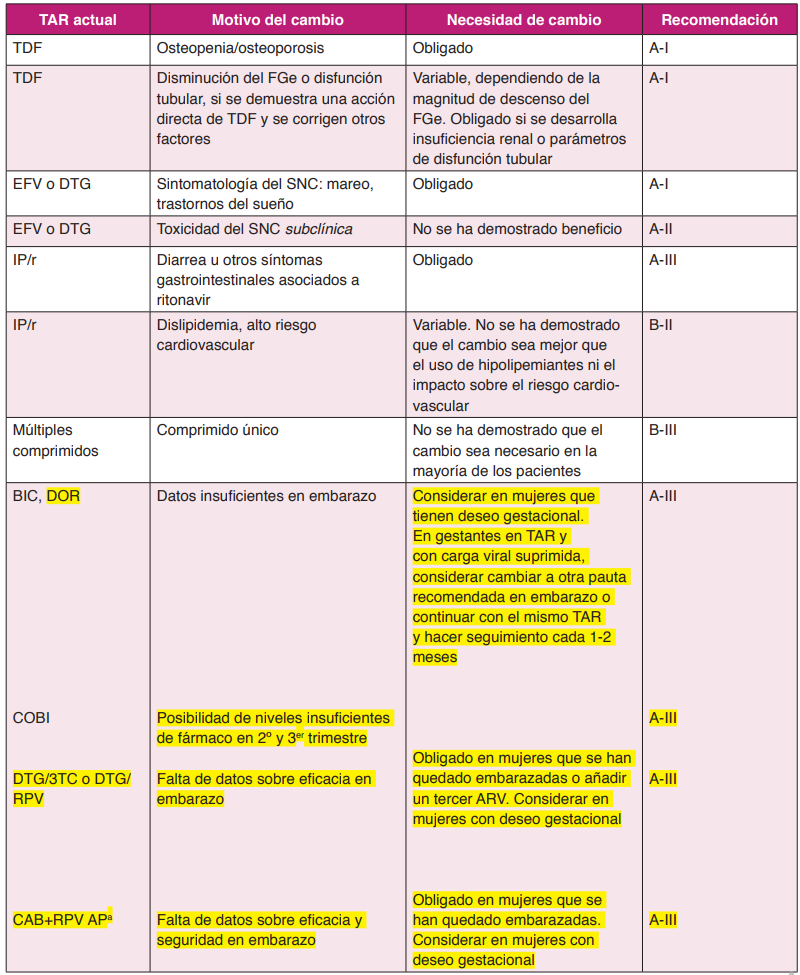
a CAB+RPV AP ha sido aprobada por la EMA, aunque en el momento de redactar estas guías aún no está comercializado en España.
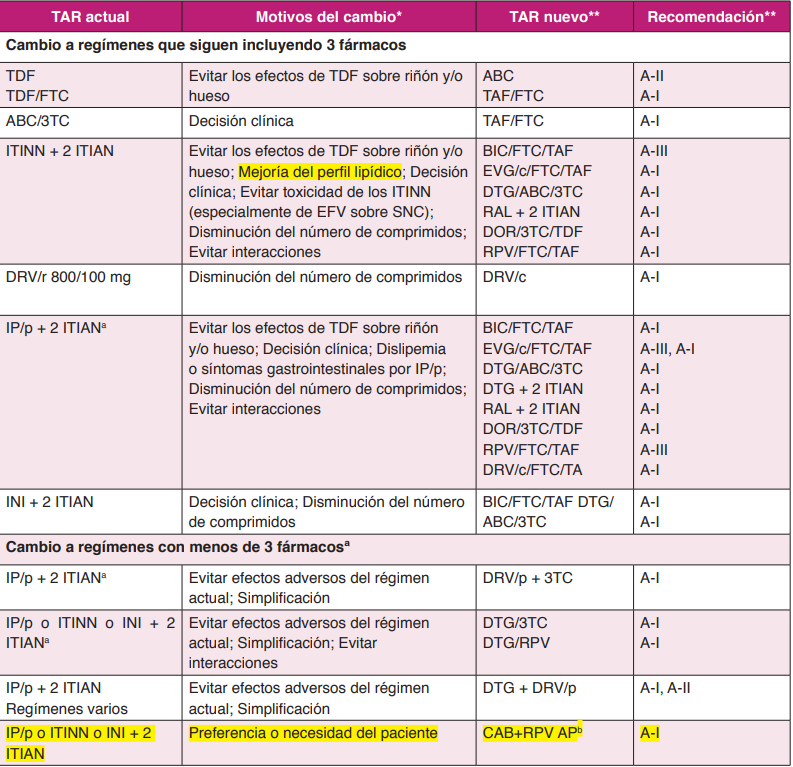
Nota:
* Véase en el texto la justificación de los motivos del cambio para cada pauta.
** Por orden alfabético dentro de cada familia (INI, ITINN, IP)
*** Véanse en el texto los criterios relativos a la fuerza de la recomendación y la base en que se sustentan.
a El cambio desde una pauta con dos ITIAN más un IP/r a dos ITIAN más un ITINN o INI de baja barrera genética solo debe hacerse si se puede garantizar la actividad antiviral de todos los fármacos de la nueva pauta.
b CAB+RPV AP ha sido aprobada por la EMA, aunque en el momento de redactar estas guías aún no está comercializada en España
Bibliografía:
Olearo F, Nguyen H, Bonnet F, Yerly S, Wandeler G, Stoeckle M, et al. Impact of the M184V/I Mutation on the Efficacy of Abacavir/Lamivudine/Dolutegravir Therapy in HIV Treatment-Experienced Patients. Open Forum Infect Dis 2019; 6:1-12.
Andreatta K, Willkom M, Martin R, et al. Switching to bictegravir/emtricitabine/tenofovir alafenamide maintained HIV-1 RNA suppression in participants with archived antiretroviral resistance including M184V/I. J Antimicrob Chemother 2019; 74:3555-6.
Jary A, Marcelin AG, Charpentier C, Wirden M, Lê MP, Peytavin G, et al. M184V/I does not impact the efficacy of abacavir/lamivudine/dolutegravir use as switch therapy in virologically suppressed patients. J Antimicrob Chemother 2020; 75:1290-3.
Mican R, de Gea A, BuscaC et al. Efficacy of switching to bictegravir/emtricitabine/ tenofovir alafenamide in patients with pre-existing nrti resistances: real world data. 18th European AIDS Conference. London 2021.
Sax PE, Meyers JL, Mugavero M, et al. Adherence to antiretroviral treatment and correlation with risk of hospitalization among commercially insured HIV patients in the United States. PLoS One 2012; 7:e31591.
Campo R, DeJesus E, Bredeek UF, et al. SWIFT: prospective 48-week study to evaluate effcacy and safety of switching to emtricitabine/tenofovir from lamivudine/abacavir in virologically suppressed HIV- 1 infected patients on a boosted protease inhibitor containing antiretroviral regimen. Clin Infect Dis 2013; 56:1637-45.
Winston A, Post FA, DeJesus E, et al. Tenofovir alafenamide plus emtricitabine versus abacavir plus lamivudine for treatment of virologically suppressed HIV-1-infected adults: a randomised, double-blind, active-controlled, non-inferiority phase 3 trial. Lancet HIV 2018; 5:e162-e171.
Negredo E, Domingo P, Perez-Alvarez N, et al. Improvement in bone mineral density after switching from tenofovir to abacavir in HIV-1-infected patients with low bone mineral density: two-centre randomized pilot study (OsteoTDF study). J Antimicrob Chemother 2014;69:3368-71.
Gallant JE, Daar ES, Raff F, et al. Effcacy and safety of tenofovir alafenamide versus tenofovir disoproxil fumarate given as fixed-dose combinations containing emtricitabine as backbones for treatment of HIV- 1 infection in virologically suppressed adults: a randomised, double-blind, active-controlled phase 3 trial. Lancet HIV 2016; 3:e158-65.
Orkin C, DeJesus E, Ramgopal M, et al. Switching from tenofovir disoproxil fumarate to tenofovir alafenamide coformulated with rilpivirine and emtricitabine in virally suppressed adults with HIV-1 infection: a randomised, double-blind, multicentre, phase 3b, non- inferiority study. Lancet HIV 2017; 4:e195-e204.
Agarwal K, Brunetto M, Seto WK, et al. 96 weeks treatment of tenofovir alafenamide vs. tenofovir disoproxil fumarate for hepatitis B virus infection. J Hepatol 2018; 68:672-81.
Mills AM, Cohen C, Dejesus E, et al. Effcacy and safety 48 weeks after switching from efavirenz to rilpivirine using emtricitabine/ tenofovir disoproxil fumarate-based single- tablet regimens. HIV Clin Trials 2013; 14:216-23.
DeJesus E, Ramgopal M, Crofoot G, et al. Switching from efavirenz, emtricitabine, and tenofovir disoproxil fumarate to tenofovir alafenamide coformulated with rilpivirine and emtricitabine in virally suppressed adults with HIV-1 infection: a randomised, double-blind, multicentre, phase 3b, non-inferiority study. Lancet HIV 2017; 4:e205-e213.
Johnson M, Kumar P, Molina JM, et al. Switching to Doravirine/Lamivudine/Tenofovir Disoproxil Fumarate (DOR/3TC/TDF) maintains HIV-1 virologic suppression through 48 Weeks: Results of the DRIVE-SHIFT Trial. J Acquir Immune Defc Syndr 2019; 81:463-72.
Nelson M, Winston A, Hill A, Mngqibisa R, Bassa A, Orkin C, et al. Efficacy, safety and central nervous system effects after switch from efavirenz/tenofovir/emtricitabine to doravirine/tenofovir/lamivudine. AIDS 2021; 35:759–67.
Palella FJ, Jr., Fisher M, Tebas P, et al. Simplifcation to rilpivirine/emtricitabine/tenofovir disoproxil fumarate from ritonavir-boosted protease inhibitor antiretroviral therapy in a randomized trial of HIV-1 RNA-suppressed participants. AIDS 2014; 28:335-44.
Tashima K, Crofoot G, Tomaka FL, et al. Cobicistat-boosted darunavir in HIV-1-infected adults: week 48 results of a Phase IIIb, open-label single-arm trial. AIDS Res Ther 2014; 11:39.
Martínez E, Negredo E, Knobel H, et al. Factors associated with the number of drugs in darunavir/ cobicistat regimens. J Antimicrob Chemother 2020; 75:208-14.
Echeverría P, Bonjoch A, Puig J, et al. Significant improvement in triglyceride levels after switching from ritonavir to cobicistat in suppressed HIV-1-infected subjects with dyslipidaemia. HIV Med 2017; 18:782- 86.
Orkin C, Molina JM, Negredo E, et al. Effcacy and safety of switching from boosted protease inhibitors plus emtricitabine and tenofovir disoproxil fumarate regimens to single- tablet darunavir, cobicistat, emtricitabine, and tenofovir alafenamide at 48 weeks in adults with virologically suppressed HIV-1 (EMERALD): a phase 3, randomised, non-inferiority trial. Lancet HIV 2018; 5:e23-e34.
Nguyen A, Calmy A, Delhumeau C, et al. A randomized cross-over study to compare raltegravir and efavirenz (SWITCH-ER study). AIDS 2011; 25:1481-87.
Eron JJ, Young B, Cooper DA, et al. Switch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicentre, double-blind, randomised controlled trials. Lancet 2010; 375:396-407.
Martinez E, Larrousse M, Llibre JM, et al. Substitution of raltegravir for ritonavir-boosted protease inhibitors in HIV-infected patients: the SPIRAL study. AIDS 2010; 24:1697-1707.
Trottier B, Lake JE, Logue K, et al. Dolutegravir/abacavir/lamivudine versus current ART in virally suppressed patients (STRIIVING): a 48-week, randomized, non-inferiority, open-label, Phase IIIb study. Antivir Ther 2017; 22:295-305.
Gatell JM, Assoumou L, Moyle G, et al. Immediate vs. deferred switching from a boosted protease inhibitor based regimen to a dolutegravir based regimen in virologically suppressed patients with high cardiovascular risk or age >50 years: Final 96 weeks results of NEAT 022 study. Clin Infect Dis 2019; 68:597-606.
Daar ES, DeJesus E, Ruane P, et al. Efficacy and safety of switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from boosted protease inhibitor-based regimens in virologically suppressed adults with HIV-1: 48 week results of a randomised, open-label, multicentre, phase 3, non- inferiority trial. Lancet HIV 2018; 5:e347-e356.
Molina JM, Ward D, Brar I, et al. Switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from dolutegravir plus abacavir and lamivudine in virologically suppressed adults with HIV- 1: 48 week results of a randomised, double-blind, multicentre, active-controlled, phase 3, non-inferiority trial. Lancet HIV 2018; 5:e357-e365.
Kityo C, Hagins D, Koenig E, et al. Switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide (B/F/TAF) in virologically suppressed HIV-1 Infected women: A randomized, open-label, multicenter, active-controlled, phase 3, noninferiority trial. J Acquir Immune Defc Syndr 2019; 82:321-28.
Acosta RK, Willkom M, Andreatta K, Liu H, Martin R, Parvangada A, et al. Switching to Bictegravir/Emtricitabine/Tenofovir Alafenamide (B/F/TAF) From Dolutegravir (DTG)+F/ TAF or DTG+F/Tenofovir Disoproxil Fumarate (TDF) in the Presence of Pre-existing NRTI Resistance. J Acquir Immune Defic Syndr 2020; 85:363-71.
Perez-Molina JA, Pulido F, Di Gianbenedetto S, et al. Individual patient data meta- analysis of randomized controlled trials of dual therapy with a boosted protease inhibitor plus lamivudine for maintenance of virological suppression: GeSIDA study 9717. J Antimicrob Chemother 2018; 73:2927-35.
Llibre JM, Hung CC, Brinson C, et al. Effcacy, safety, and tolerability of dolutegravir- rilpivirine for the maintenance of virological suppression in adults with HIV-1: phase 3, randomised, non-inferiority SWORD-1 and SWORD-2 studies. Lancet 2018; 391:839-49.
van Wyk J, Orkin C, Rubio R, Bogner J, Baker D, Khuong-Josses M-A, et al. Brief Report: Durable Suppression and Low Rate of Virologic Failure 3 Years After Switch to Dolutegravir + Rilpivirine 2-Drug Regimen: 148-Week Results From the SWORD-1 and SWORD-2 Randomized Clinical Trials. J Acquir Immune Defic Syndr 2020; 85:325-30.
van Wyk J, Ajana F, Bisshop F, De Wit S, Osiyemi O, Portilla Sogorb J, et al. Efficacy and Safety of Switching to Dolutegravir/Lamivudine Fixed-Dose 2-Drug Regimen vs Continuing a Tenofovir Alafenamide-Based 3- or 4-Drug Regimen for Maintenance of Virologic Suppression in Adults Living with Human Immunodeficiency Virus Type 1: Phase 3, Randomized, Noninferiority TANGO Study. Clin Infect Dis 2020; 71:1920-9.
Llibre J, Alves C, Cheng CY, et al. Switching to the 2-drug regimen of dolutegravir/ lamivudine (DTG/3TC) fixed-dose combination (FDC) is non-inferior to continuing a 3-drug regimen through 48 weeks in a randomized clinical trial (SALSA). IAS 2021, 11th IAS Conference on HIV Science, July 18-21, 2021. Abstract OALB0303.
van Wyk J, Ait-Khaled M, Santos J, Scholten S, Wohlfeiler M, Ajana F, et al. Brief Report: Improvement in Metabolic Health Parameters at Week 48 After Switching From a Tenofovir Alafenamide-Based 3- or 4-Drug Regimen to the 2-Drug Regimen of Dolutegravir/ Lamivudine: The TANGO Study. J Acquir Immune Defic Syndr 2021; 87:794-800.
Spinner CD, Kümmerle T, Schneider J, Cordes C, Heiken H, Stellbrink H-J, et al. Efficacy and Safety of Switching to Dolutegravir With Boosted Darunavir in Virologically Suppressed Adults With HIV-1: A Randomized, Open-Label, Multicenter, Phase 3, Noninferiority Trial: The DUALIS Study. Open Forum Infect Dis 2020; 7:1-8.
Wolf E, Boesecke C, Balogh A, Bidner H, Cordes C, Heiken H, et al. Virologic Outcomes by Resistance Category and Pretreatment in the DUALIS Study. Conference on Retrovirus and Opportunistic Infections. March 8-11. Boston, Massachusetts, 2020.
Rial-Crestelo D, Pinto-Martínez A, Pulido F. Cabotegravir and rilpivirine for the treatment of HIV. Expert Rev Anti Infect Ther 2020; 18:393-404.
Swindells S, Andrade-Villanueva JF, Richmond GJ, Rizzardini G, Baumgarten A, Masiá M, et al. Long-acting cabotegravir and rilpivirine for maintenance of HIV-1 suppression. N Engl J Med 2020; 382:1112-23.
Overton ET, Richmond G, Rizzardini G, Jaeger H, Orrell C, Nagimova F, et al. Long- acting cabotegravir and rilpivirine dosed every 2 months in adults with HIV-1 infection (ATLAS-2M), 48-week results: a randomised, multicentre, open-label, phase 3b, non- inferiority study. Lancet 2020;396:1994–2005.
Swindells S, Lutz T, van Zyl L, Porteiro N, Stoll M, Mitha E, et al. Long-acting cabotegravir + rilpivirine for HIV-1 treatment. AIDS 2021;Publish Ahead of print. doi. org/10.1097/QAD.0000000000003025.
Week 96 Efficacy and Safety of Cabotegravir + Rilpivirine Every 2 Months: Atlas-2M. Conference on Retroviruses and Opportunistic Infections. Boston USA. March 6-10, 2021.
Orkin C, Arasteh K, Górgolas Hernández-Mora M, Pokrovsky V, Overton ET, Girard P-M, et al. Long-Acting Cabotegravir and Rilpivirine after Oral Induction for HIV-1 Infection. N Engl J Med 2020; 382:1124-35.
Orkin C, Oka S, Philibert P, Brinson C, Bassa A, Gusev D, et al. Long-acting cabotegravir plus rilpivirine for treatment in adults with HIV-1 infection: 96-week results of the randomised, open-label, phase 3 FLAIR study. Lancet HIV 2021; 8:e185–96.v
Cutrell AG, Schapiro JM, Perno CF, Kuritzkes DR, Quercia R, Patel P, et al. Exploring predictors of HIV-1 virologic failure to long-acting cabotegravir and rilpivirine: a multivariable analysis. AIDS 2021; 35:1333-42.
5. FRACASO DEL TRATAMIENTO ANTIRRETROVIRAL
5.1. Definiciones
5.2. Incidencia y factores determinantes del FV
5.3. Objetivo del tratamiento tras FV
5.4. Estrategias para mejorar el éxito de los tratamientos de rescate
5.5. Escenarios clínicos de FV
Definiciones
- Fracaso virológico (FV): CVP >200 cop/mL transcurridas 24 semanas desde el inicio del TAR, confirmada en una segunda muestra consecutiva. A veces, pueden ser necesarias más de 24 semanas de TAR para alcanzar la CVP <50 cop/mL, particularmente si la CVP basal es elevada (>5 log VIH-ARN cop/mL), en regímenes que no contengan un INI o en determinados subtipos genéticos (i.e. subtipo F*).
- Repuntes virológicos transitorios aislados (“blips”): valores de CVP entre 50 y 1000 cop/mL, con valores de CVP previa y posterior <50 cop/mL. Los “blips” aislados con CVP <200 cop/mL no parecen tener repercusión clínica. Sin embargo, los “blips” frecuentes o con cifras de CVP> 200 cop/mL se han asociado a mayor riesgo de FV y aparición de MR12.
- Viremia de bajo nivel (VBN): CVP 50-1000 cop/mL en al menos dos muestras Estas viremias se pueden dividir en dos subgrupos: (i) viremias de muy bajo nivel (VMBN) CVP 50-200 cop/mL; y (ii) viremias de moderado bajo nivel (VMoBN) CVP 200-1000 cop/mL. Si bien existen datos contradictorios, en general en VMBN no parece haber evolución viral con selección de MR, ni incremento de la CVP a viremias de >500 cop/mL, las VMoBN se han asociado de forma más constante a FV con viremias de >500 cop/mL y selección de MR.
Incidencia y factores determinantes del FV
Las tasas de FV de los TAR de inicio preferentes a las 48 semanas son inferiores al 10%. Los factores que influyen en el FV son:
- Mala adherencia al tratamiento o al seguimiento de controles médicos.
- Efectos adversos
- Interacciones farmacocinéticas con fármacos, productos de herboristería, alimentos, complementos nutricionales o drogas de recreo
- Pre-existencia de MR.
En ausencia de MR pre-existentes, la baja barrera a la selección de resistencias de los FAR no conlleva un mayor riesgo de FV en sí.
Objetivo del tratamiento tras FV
El objetivo de la pauta de rescate es conseguir de nuevo la supresión viral (<50 cop/ mL). Para ello, debe instaurarse un TAR con dos fármacos con actividad plena de los que al menos uno de ellos debe tener una alta barrera a las resistencias (INI de segunda generación o DRV/p) , incluyendo, si es posible, al menos un FAR de una familia no usada previamente. El inicio de un TAR de rescate no debe retrasarse para evitar el acúmulo de MR y el deterioro inmunológico y clínico del paciente.
Estrategias para mejorar el éxito de los tratamientos de rescate
- Facilitar la adherencia y tolerabilidad al TAR. Corregir causas de mala adherencia, posibles errores de dosificación e interacciones medicamentosas; pautar un TAR bien tolerado, lo más simple posible y adaptado al estilo de vida y las comorbilidades del paciente.
- Pruebas de resistencia. El FV a menudo se asocia a la acumulación de MR que limitan la eficacia de los TAR. Dichas MR no siempre se pueden predecir a partir de la historia clínica3. La prueba debe realizarse en plasma, idealmente mientras el paciente recibe el TAR al que está fracasando, o no más de 4 semanas tras la suspensión del mismo, y con una CVP preferiblemente >200 cop/mL. A mayor CVP más probabilidades hay de secuenciación exitosa del virus4. Aunque existen tecnologías de secuenciación ultrasensibles, a día de hoy únicamente se deben considerar aquellas MR presentes en al menos un 15% de la población viral (punto de corte equivalente al de la secuenciación Sanger) para modificar el TAR. Sólo si se incluyen fármacos que no tengan una alta barrera a las resistencias en el rescate, podría ser clínicamente relevante considerar MR presentes entre el 5% y el 15% de los virus5. Las plataformas de secuenciación masiva detectan resistencias seleccionadas en poblaciones virales minoritarias pero no aumentan su sensibilidad en la secuenciación de VBN.
Se debe asumir que las MR no desaparecen, por lo que hay que considerar todas las MR acumuladas en todas las pruebas de resistencias de las que se disponga a la hora de diseñar un nuevo TAR. Además de estas MR, hay que considerar las que se hayan podido seleccionar tras FV en los que no se disponga de estudios de resistencias.
Aunque ya existen estudios en los que se ha utilizado la determinación de resistencias en DNA proviral en pacientes suprimidos o con VBN, su uso en la rutina es aún muy limitado. La escasez de test comercializados (actualmente hay sólo uno aprobado por la FDA para secuenciación de CVP >500 cop/mL GenoSure® MG) -que hacen que la mayoría de los test sean home made-, y la necesidad de expertos que sean capaces de leer correctamente los resultados están haciendo que su implementación en la rutina sea más lenta de lo esperado678. Sin embargo, consideramos que interpretados con precaución estos estudios pueden ayudar a detectar mutaciones archivadas en individuos con VBN o CVP indetectable.
c. Tropismo viral. Debe determinarse cuando se plantea el uso de MVC. Una vez se han identificado cepas con tropismo no-R5, no debe de volver a plantearse un tratamiento con un antagonista de CCR5.
d. Monitorización de la concentración plasmática de fármacos. No se ha demostrado que su utilización mejore la eficacia del TAR de rescate, por lo que no debe usarse en rutina clínica, excepto para ajustar dosis si existen interacciones farmacológicas; en pacientes con peso extremo o insuficiencia hepática o renal; en pacientes con dudas en relación con la adherencia o VBN a pesar de referir una buena adherencia9.
Recomendaciones generales ante un FV
- Se deben analizar las causas que motivaron el fracaso, la historia farmacológica y los fracasos previos. El nuevo TAR debe ser lo más cómodo y bien tolerado posible (A-III).
- El cambio del TAR por FV debe efectuarse precozmente para evitar la acumulación de MR y facilitar la respuesta al nuevo tratamiento (A-III).
- Para confeccionar un régimen de rescate óptimo, se debe realizar un estudio de resistencias en plasma y el tropismo viral si se plantea el uso de un antagonista de CCR5 y no se ha descartado previamente la presencia de variantes no-R5. Deben de valorarse todas las MR detectadas en genotipos realizados previamente y posibles MR seleccionadas en FV previos no secuenciados (A-I).
- El nuevo TAR debe contener 2 FAR completamente activos, si al menos uno de ellos tiene una alta barrera a las resistencias (DRV/r o INI de segunda generación). En caso de que el TAR no incluya un FAR de alta barrera a las resistencias, deberá incluir 3 fármacos plenamente activos (A-I).
- En los pacientes con VHB que se modifique el TAR por FV debemos asegurarnos de que el VHB quede cubierto, para evitar rebrotes de replicación del mismo (A-I).
Escenarios clínicos de FV
5.5.1 FV CON VIREMIAS DE BAJO NIVEL
Bajo este epígrafe se consideran 2 situaciones:
a) Viremias de muy bajo nivel (VMBN): CVP de 50-200 cop/mL
Existe discusión sobre la actitud terapéutica más adecuada en esta situación,
porque:
• Las pruebas que utilizan técnicas de PCR en tiempo real (TaqMan® o Abbott RealTime®) presentan mayor sensibilidad para detectar VMBN4
, siendo más susceptibles a falsos positivos técnicos.
La VMBN persistente puede ocasionarse a partir de la integración del VIH en una región transcripcionalmente activa de una población de linfocitos CD4+ clonalmente expandida, sin que exista replicación de virus competentes10. En estas viremias no se ha visto que haya evolución viral ni selección de nuevas MR.
• Es difícil amplificar material genómico suficiente para realizar una prueba genotípica de MR en plasma, aunque se puede aumentar un poco la
sensibilidad del test concentrando el virus a partir de un volumen mayor de plasma (3 ml)7. A pesar de sus limitaciones8, los estudios genotípicos en ADN proviral pueden ser útiles en este escenario cuando son evaluados por expertos.
b) Viremias de moderado bajo nivel (VMoBN): CVP: 200-1000 cop/mL
La presencia de CVP de 200-1000 cop/mL se asocia a un mayor riesgo de FV111213 con selección de MR9, por lo que deben considerarse como FV. Ante esta situación es preciso un estudio genotípico y elaborar una pauta de rescate teniendo en cuenta las MR, los FV previos, los problemas de adherencia, la toxicidad, el riesgo de interacciones y la comodidad de los FAR.
Recomendaciones en viremias de bajo nivel (VBN)
- En este escenario puede ser recomendable un seguimiento clínico-analítico más estrecho (trimestral) (C-III).
- En ausencia de evidencia sólida, si el estudio genotípico no muestra MR, se recomienda administrar un TAR con alta barrera frente al desarrollo de resistencias (en especial en el escenario de VMoBN), reforzar la adherencia y revisar potenciales interacciones (A-III).
- Si, por lo contrario, el test muestra MR, se recomienda actuar como si se tratara de un FV “clásico”, optimizando el tratamiento según el perfil de MR (A-I).
- No se recomienda la intensificación terapéutica añadiendo un solo fármaco activo (A-III).
5.5.2. PRIMER FV
Es el fracaso a la primera línea de TAR. La selección de MR y las pautas de segunda línea difieren dependiendo de la pauta de inicio utilizada.
- FV a 2 ITIAN+INI. Múltiples ensayos clínicos demuestran que en pacientes que inician regímenes triples con DTG o BIC siendo naïve a los antirretrovirales y sin MR transmitidas, el FV es muy raro y, salvo algún caso anecdótico, no se asocia a MR en la integrasa ni en la transcriptasa inversa. Sin embargo, la experiencia fuera de ensayos clínicos puede ser distinta, y en situaciones de baja adherencia o interacciones medicamentosas (con tuberculostáticos, por ejemplo), puede desarrollarse EVG, suele asociarse a MR con resistencia a ITIANe INI14. Por el contrario, el FV a RAL o EVG, suele asociarse a MR cruzadas a INI, a menudo acompañadas de MR a ITIAN 15. No existen estudios que evalúen la mejor estrategia tras el FV a DTG o BIC porque son fármacos recientes y se asocian a tasas muy bajas de FV. En el caso de FV a INI de primera generación la pauta de rescate se decidirá en base a los resultados del estudio de resistencias. En el caso de incluir DTG, este se administrará a dosis de 50 mg BID (aunque no se identifiquen MR a los INI).
- FV a IP/p + 2 ITIAN. En los pacientes que fracasan a una pauta inicial con IP/p + 2 ITIAN, el FV se debe habitualmente a mala adherencia. La probabilidad de seleccionar MR a IP/p en pacientes naïve a los ARV sin MR transmitidas es prácticamente nula. Además, los IP/p protegen frente a la selección de MR a los ITIAN acompañantes, observándose sólo, y muy raramente, la M184I/V. Una revisión sistemática en pacientes con fracaso a una pauta de inicio con IP/p + 2 ITIAN, sin MR, demostró que mantener el mismo TAR es igual de eficaz que cambiar a una pauta de rescate con FAR de otras familias, especialmente en personas con cifras elevadas de linfocitos CD4+16.
- FV a 2 ITIAN+ITINN. El FV a EFV o NVP frecuentemente se asocia al desarrollo de MR, siendo las más frecuentes: K103N, , G190A, L100I o Y181C. El FV a RPV selecciona preferentemente las MR E138K y Y181C, que confieren resistencia cruzada a todos los ITINN (incluso resistencia de bajo nivel a DOR si aparecen conjuntamente). La mutación K103N aislada no confiere resistencias a RPV, ETV ni DOR. EL FV a DOR selecciona preferentemente las mutaciones V106I y F227C, así como A98G. El FV a ITINN suele acompañarse también de MR a ITIAN, especialmente M184V/I y, con menor frecuencia, K65R. El estudio DAWNING17 demostró que en pacientes que fracasan a ITINN, el cambio a DTG es superior al cambio a un régimen con LPV/r, incluso en presencia de MR a uno de los ITIAN. Datos recientes del estudio NADIA confirman los buenos resultados de DTG+2ITIAN en este escenario de fracaso y muestran una eficacia similar en este rescate a la combinación de DRV/p más 2 ITIAN. En este mismo escenario - no FV a INI especialmente si sólo está presente una M184V en la RT-, asumiendo la similar barrera a las resistencias de DTG vs BIC y la potencial mayor barrera a las resistencia de TAF vs TDF la combinación TAF/FTC/BIC podría ser también activa. Tres ensayos clínicos demostraron la no inferioridad de la biterapia con LPV/r + RAL frente a LPV/r + 2 o 3 ITIAN181920 en esta situación clínica.
Recomendaciones en primer FV:
- El diseño del TAR tras un FV dependerá del resultado del test de resistencias. Si existen MR, se adecuarán los cambios de TAR a las mismas, con el objetivo de conseguir un TAR de rescate plenamente activo (A-II).
- En ausencia de MR, se deberá descartar mala adherencia y se investigarán posibles interacciones, errores en la prescripción o la toma, y otros motivos en la vida del paciente que dificulten una correcta toma de la medicación (A-II). Una vez corregidos, se podrá continuar el mismo TAR con un control precoz (1-3 meses dependiendo de la CVP) de la CVP (C-III). Alternativamente- especialmente en FV a esquemas con baja barrera a las resistencias- se modificará el TAR según lo resumido en la (Tabla 6) (A-I).
- Los pacientes que desarrollen FV a un régimen con ITINN podrán:
- Cambiar a un TAR con 2 ITIAN+DTG o DRV/p (A-I). En este escenario el rescate se podría hacer ocasionalmente con las coformulaciones TAF/FTC/DRV/c o TAF/FTC/BIC (C-III), aunque no existen datos ni está aprobada actualmente su indicación si hay resistencia previa a alguno de sus componentes.
- En presencia de MR a 1 ITIAN, es preferible optar por un régimen con DTG o con DRV/p que con LPV/r (A-I).
- Actualmente se están evaluando regímenes que combinan DTG+DRV/c, posiblemente más seguros, convenientes y mejor tolerados que LPV/r + RAL (B-II).
- Los pacientes que desarrollen FV a una pauta inicial con IP/p + 2 ITIAN,
podrán:
- el mismo TAR sin no ha seleccionado MR (A-I).
- Cambiar a un esquema con DTG o probablemente TAF/FTC/BIC -si no FV previo a INI y actividad plena de TAF- , sobre todo si no existen MR en
la integrasa y hay problemas de tolerancia al IP/p (B-III).
- En pacientes que desarrollen FV a una pauta inicial con 2 ITIAN + DTG o
BIC
- Si el genotipado muestra MR, construir pauta de rescate siguiendo las recomendaciones generales ante un FV del punto 5.4 (A-I).
- Se valorará, si se confirma el FV y descartan otras causas del mismo, cambiar a la coformulación TAF/FTC/DRV/c (C-III).
- Si se decide mantener DTG se administrará doble dosis mientras exista al menos susceptibilidad residual a este fármaco (A-III).
5.5.3. FV AVANZADO
Es el FV a la segunda o sucesivas líneas de TAR. En este escenario, la mayoría de los pacientes pueden presentan MR a dos o más familias de FAR. Los principales ensayos clínicos de pautas de tratamientos de rescate avanzado se resumen en la (Tabla 6).
a. Inhibidores de la Los IP/p son los fármacos que han demostrado tener una mayor barrera a las resistencias y de entre ellos DRV/p por farmacocinética (mayor cociente inhibitorio) y mayor constante de disociación. Cuando existe alguna mutación mayor de resistencia a DRV, se recomienda utilizar DRV/r 600/100 mg BID21-25.
b. La eficacia residual de los ITIAN en presencia de MR es motivo de debate. El mantenimiento de XTC en presencia de M184V hipersensibiliza el virus a TDF/ TAF y AZT (si no hay TAMs) o revierte parcialmente su sensibilidad en presencia de TAMs, reduce el fitness viral y contribuye a disminuir la CVP en 0,5 log, sin apenas toxicidad ni coste añadido, Los estudios EARNEST y SECOND LINE 1819 demostraron no sólo la persistencia de efectividad de los ITIAN; además, la presencia de MR a ITIAN se asociaba a una mejor respuesta a un régimen basado en LPV/r cuando éste se combinaba con 2 ITIANs. En comparación, la respuesta a monoterapia con LPV/r fue significativamente peor.
c. Inhibidores de la integrasa. La presencia de la mutación Q148H/R en la integrasa asociada con otras MR adicionales se relacionó con una menor eficacia de DTG262728. Asimismo, la respuesta a DTG disminuye en presencia en presencia de N155H/S147G y dos MR secundarias30. BIC tiene un perfil de resistencia muy similar al de DTG. Hasta la fecha no ha sido investigado como TAR de rescate y siempre se presenta co-formulado con TAF/FTC.
En pacientes naïve a INI, con CVP >1.000 cop/mL y resistencia a ≥2 familias de FAR (estudio SAILING)26, DTG 50 mg QD resultó superior a RAL, ambos con terapia optimizada. La selección de resistencias fue significativamente menor con DTG tanto en el gen de la integrasa como frente a los ITIAN.
En caso de FV previo a INI, con o sin mutaciones en el gen de la IN, se deberá utilizar DTG 50 mg BID en caso de que se considere su utilización tras analizar el total de las MR29.
Recomendaciones en FV avanzado:
- El diseño de un tratamiento de rescate deberá guiarse siempre por los resultados de un test de resistencias obtenido en el momento del FV, teniendo en cuenta los genotipos acumulados previamente así como las pautas de tratamiento previo y las MR no recogidas en los estudios previosque pudiesen estar presentes (A-I).
- Todo TAR de rescate avanzado incluirá DRV/p (la pauta será de DRV/r 600/100 mg BID en caso de que se identifiquen MR del score específico de DRV) en caso de que el estudio de resistencias muestre algún nivel de actividad (A-I).
- DRV/p deberá acompañarse de otros FAR (ITIAN, ITIANN e INI) hasta conseguir un régimen plenamente activo (A-I).
- En caso de que con los ITIAN, ITIANN, IP e INI, no se pueda construir un esquema con garantías de supresión virológica completa y sostenida se deberá incluir en el tratamiento de rescate un fármaco que actúe en otras dianas virales (inhibidor de fusión, inhibidor del acoplamiento, ibalizumab, inhibidor de la cápside o inhibidor de la traslocación de la transcriptasa inversa).
- Algunos autores recomiendan mantener XTC incluso en presencia de M184V (B-III).
- El INI de elección para TAR de rescate es DTG, que se administrará QD o BID según el patrón de mutaciones en la integrasa y la exposición previa o no a INI (A-I). La posibilidad de utilizar BIC, dada su coformulación obligada con TAF/FTC, estará condicionada por la ausencia de MR/FV a INI y la presencia de actividad plena de TAF.
5.5.4. FV EN PACIENTES CON ESCASAS OPCIONES TERAPÉUTICAS
Existen datos de nuevos fármacos que actúan en diferentes dianas del virus:
- Fostemsavir313233 (GSK3684934; anteriormente BMS-663068): inhibidor del acoplamiento (attachment) y prodroga del Temsavir que se administra a dosis de 1 comp de 600 mg BID ha demostrado en el estudio BRIGHTE una tasa de respuesta (CVP <40 cop/mL) en la semana 96 del 60% en la cohorte randomizada (CR: al menos 1 fármaco activo) y del 37% en la cohorte no randomizada (NR: no necesidad de fármacos activos). Con una mediana basal de 80 linfocitos CD4/μL (99 para la CR y 46 para la NR), a 96 semanas la recuperación media fue 205 células/μL en la CR y de 119 células/μL en la Un 7% discontinuaron por efectos adversos. Ha sido ya aprobado por la FDA y la EMA.
- Ibalizumab34: anticuerpo monoclonal humanizado que se une al segundo dominio extracelular del receptor CD4, bloqueándolo. No tiene resistencia cruzada con otros FAR ni interacciones farmacocinéticas significativas y ha demostrado eficacia virológica en pacientes severamente pretratados; el 43% de los pacientes alcanzaron CV<50 cop/mL en la semana 25. Ha sido ya aprobado por la FDA y la EMA. El precio y la administración intravenosa son importantes limitaciones al uso del fármaco, que queda reservado para situaciones en la que la alternativas no son apropiadas.
- El inhibidor de la cápside (Lenacapavir, GS-620735, Gilead Sciences) ,cuya autorización para el tratamiento de pacientes extensamente pretratados ha sido recientemente solicitada a la FDA (Junio 2021) y EMA (agosto 2021) en base a los datos del estudio CAPELLA que se presentaron en EACS 2021, supone una nueva opción terapéutica altamente prometedora que pronto estará disponible para pacientes en fracaso terapéutico avanzado.
- El inhibidor de la traslocación de la transcriptasa inversa Islatravir (MK-8591, MSD)36, actualmente en evaluación administrado junto a DOR en pacientes extensamente pretratados en un estudio en fase III (MK-8591A-019), aparece como una alternativa terapéutica igualmente prometedora.
Recomendaciones en pacientes sin opciones terapéuticas:
- Se recomienda derivar al paciente a un centro con experiencia y acceso a nuevos FAR a través de ensayos o programas de acceso expandido, que puedan estar disponibles (A-III).
- En el tratamiento de rescate de individuos altamente tratados y con escasas opciones terapéuticas, se deberán intentar incluir fármacos de nuevas familias y seleccionar los fármacos con mayor actividad para con ellos diseñar la mejor opción de rescate posible (C-III).
- Si no es posible construir un TAR de rescate efectivo con al menos dos FAR activos, si la situación clinicoinmunológica del paciente lo permite, debe intentarse no exponer al paciente a monoterapia funcional con un solo FAR por el elevado riesgo de FV y selección de MR a ese único FAR (A-III).
- Se recomienda esperar a la aparición de nuevo FAR adicional que permita construir un esquema supresor. Mientras, se construirá un tratamiento “puente” que combine efectividad residual de los fármacos y permita una supresión parcial de la replicación viral, minimizando la acumulación de MR que limiten la efectividad de futuros fármacos. El objetivo de este TAR “puente” es retrasar la progresión clínica, el deterioro inmunológico y limitar la acumulación de MR (A-III).
- En cuanto sea posible, este tratamiento debe cambiarse a un TAR supresor con 2-3 FAR activos que incluya fármacos de nuevas familias (A-III).
- Jamás debe interrumpirse el TAR, ya que el descenso de la cifra de linfocitos CD4+ es mayor que si se continúa administrando un TAR no supresor37 (A-I).
Tablas:
(las referencias bibliográficas corresponden a las del capítulo 5: Fracaso del tratamiento antirretroviral)
Bibliografía:
Lee PK, Kieffer TL, Siliciano RF, et al. HIV-1 viral load blips are of limited clinical significance. J Antimicrob Chemother 2006; 57:803-5.
Grennan JT, Loutfy MR, Su D, et al. Magnitude of virologic blips is associated with a higher risk for virologic rebound in HIV-infected individuals: a recurrent events analysis. J Infect Dis 2012; 205:1230-8.
Palella FJ, Jr., Armon C, Buchacz K, et al. The association of HIV susceptibility testing with survival among HIV-infected patients receiving antiretroviral therapy: a cohort study. Ann Intern Med 2009; 151:73-84.
Ryscavage P, Kelly S, Li , et al . Significance and clinical management of persistent low-level viremia and very-low-level viremia in HIV-1-infected patients. Antimicrob Agents Chemother. 2014;58(7):3585-98.
Li JZ, Paredes R, Ribaudo HJ, Svarovskaia ES, et al. Low-frequency HIV-1 drug resistance mutations and risk of NNRTI-based antiretroviral treatment failure: a systematic review and pooled analysis. JAMA. 2011. 305(13):1327-35
Monogram Biosciences. GenoSure Archive: HIV-1 next generation DNA sequencing assay. http://www.monogrambio.com/hiv-tests/suppression-management/genosure- archive
Parra-Ruiz J, Alvarez M, Chueca N, et al. Resistencias genotipicas en pacientes con VIH-1 y grados de viremia persistentemente bajos. Enferm Infecc Microbiol Clin 2009; 27:75-80.
Sotillo A, Sierra O, Martínez-Prats L, et al. Analysis of drug resistance mutations in whole blood DNA from HIV-1 infected patients by single genome and ultradeep sequencing analysis. J Virol Methods 2018; 260:1-5.
Back DJ, Khoo SH, Gibbons SE, Merry C. The role of therapeutic drug monitoring in treatment of HIV infection. Br J Clin Pharmacol. 2001 ;51(4):301-8
Jacobs JL, Halvas EK, Tosiano MA Mellors JW. Persistent HIV-1 viremia on antiretroviral therapy: measurement and mechanisms. Front Microbiol 2019; 10: 2383.
Laprise C, de Pokomandy A, Baril JG, et al. Virologic failure following persistent low- level viremia in a cohort of HIV-positive patients: results from 12 years of observation. Clin Infect Dis 2013; 57:1489-96.
Vandenhende MA, Perrier A, Bonnet F, et al. Risk of virological failure in HIV-1-infected patients experiencing low-level viraemia under active antiretroviral therapy (ANRS C03 cohort study). Antivir Ther 2015; 20:655-60.
Bernal E, Gómez JM, Jarrín I, et al, Low-level viremia is associated with clinical progression in HIV-infected patients receiving antiretroviral treatment. J Acquir Immune Defic Syndr 2018; 78:329-37.
Lübke N, Jensen B, Hüttig F,et al. Failure of dolutegravir first-line ART with selection of virus carrying R263K and G118R. N Engl J Med 2019; 381:887-9.
Lepik KJ, Harrigan PR, Yip B, et al. Emergent drug resistance with integrase strand transfer inhibitor-based regimens. AIDS 2017; 31:1425-34.
Zheng Y, Hughes MD, Lockman S, et al. Antiretroviral therapy and efficacy after virologic failure on first-line boosted protease inhibitor regimens. Clin Infect Dis 2014; 59:888-96.
Aboud M, Kaplan R, Lombaard J, et al. Dolutegravir versus ritonavir-boosted lopinavir both with dual nucleoside reverse transcriptase inhibitor therapy in adults with HIV-1 infection in whom first-line therapy has failed (Dawning): an open-label, non-inferiority, phase 3b trial. Lancet Infect Dis 2019; 19:253-64.
Boyd MA, Kumarasamy N, Moore CL, et al. Ritonavir-boosted lopinavir plus nucleoside or nucleotide reverse transcriptase inhibitors versus ritonavir-boosted lopinavir plus raltegravir for treatment of HIV-1 infection in adults with virological failure of a standard first-line bART regimen (SECOND-LINE): a randomised, open-label, non-inferiority study. Lancet 2013; 381:2091-9.
Paton NI, Kityo C, Hoppe A, et al. Assessment of second-line antiretroviral regimens for HIV therapy in Africa. N Engl J Med 2014; 371:234-47.
La Rosa AM, Harrison LJ, Taiwo B, et al. Raltegravir in second-line antiretroviral therapy in resource-limited settings (SELECT): a randomised, phase 3, non-inferiority study. Lancet HIV 2016; 3:e247-58.
Clotet B, Bellos N, Molina JM, et al. Efficacy and safety of darunavir-ritonavir at week 48 in treatment-experienced patients with HIV-1 infection in POWER 1 and 2: a pooled subgroup analysis of data from two randomised trials. Lancet 2007; 369:1169-78.
Madruga JV, Berger D, McMurchie M, et al. Efficacy and safety of darunavir-ritonavir compared with that of lopinavir-ritonavir at 48 weeks in treatment-experienced, HIV- infected patients in TITAN: a randomised controlled phase III trial. Lancet 2007; 370:49- 58.
Sension M, Cahn P, Domingo P, et al. Subgroup analysis of virological response rates with once- and twice-daily darunavir/ritonavir in treatment-experienced patients without darunavir resistance-associated mutations in the ODIN trial. HIV Med 2013; 14:437-44.
Cahn P, Fourie J, Grinsztejn B, et al. Week 48 analysis of once-daily vs. twice-daily darunavir/ritonavir in treatment-experienced HIV-1-infected patients. AIDS 2011; 25:929- 39.
Imaz A, del Saz SV, Ribas MA, et al. Raltegravir, etravirine, and ritonavir-boosted darunavir: a safe and successful rescue regimen for multidrug-resistant HIV-1 infection. J Acquir Immune Defic Syndr 2009; 52:382-6.
Cahn P, Pozniak AL, Mingrone H, et al. Dolutegravir versus raltegravir in antiretroviral- experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet 2013; 382:700-8.
Eron JJ, Clotet B, Durant J, et al. Safety and efficacy of dolutegravir in treatment- experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study. J Infect Dis 2013; 207:740-8.
Castagna A, Maggiolo F, Penco G, et al. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the Phase III VIKING-3 Study. J Infect Dis 2014; 210:354-62.
Akil B, Blick G, Hagins DP, et al. Dolutegravir versus placebo in subjects harbouring HIV-1 with integrase inhibitor resistance associated substitutions: 48-week results from VIKING-4, a randomized study. Antivir Ther 2015; 20:343-8.
Hardy I, Brenner B, Quashie P, et al. Evolution of a novel pathway leading to dolutegravir resistance in a patient harbouring N155H and multiclass drug resistance. J Antimicrob Chemother 2015; 70:405-11.
Thompson M, Lalerzari J, Kaplan R et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug fostemsavir in antiretroviral-experienced subjects: week 48 analysis of AI438011, a Phase IIb, randomized controlled trial. Antivir Ther 2017; 22:215-23.
Kozal M, Aberg J, Pialoux G, et al. Phase 3 study of fostemsavir in heavily treatment- experienced HIV-1–infected participants: Day 8 and week 24 primary efficacy and safety results (BRIGHTE Study, formerly 205888/AI438-047). 16th European AIDS Conference Milan, Italy, October 25-27, 2017; Abstract PS8/5.
M. Lataillade, J. Lalezari, J. Aberg, et al: Week 96 safety and efficacy of the novel HIV- 1 attachment inhibitor prodrug fostemsavir in heavily treatment-experienced participants infected with multi-drug–resistant HIV-1 (BRIGHTE Study). International AIDS Society 2019. Abstr MOAB0102
Emu B, Fessel J, Schrader S, et al. Phase 3 Study of Ibalizumab for Multidrug-Resistant HIV-1. N Engl J Med 2018; 379:645-54.
Stephen R Yant 1 , Andrew Mulato 2 , Derek Hansen, et al. A Highly Potent Long- Acting Small-Molecule HIV-1 Capsid Inhibitor With Efficacy in a Humanized Mouse Model. Nat Med, 2019; 25 (9), 1377-1384
Dirk Schürmann , Deanne Jackson Rudd, Saijuan Zhang, et al. Safety, Pharmacokinetics, and Antiretroviral Activity of Islatravir (ISL, MK-8591), a Novel Nucleoside Reverse Transcriptase Translocation Inhibitor, Following Single-Dose Administration to Treatment- Naive Adults Infected With HIV-1: An Open-Label, Phase 1b, Consecutive-Panel Trial. Lancet HIV 2020 Jan 3[Online ahead of print]
Lawrence J, Mayers D, Huppler Hullsiek K, et al. Structured Treatment Interruption in Patients with Multidrug-Resistant Human Immunodeficiency Virus. N Engl J Med 2003; 349:83.
Ruxrungtham K, Pedro RJ, Latiff GH, et al. Impact of reverse transcriptase resistance on the efficacy of TMC125 (etravirine) with two nucleoside reverse transcriptase inhibitors in protease inhibitor-naive, nonnucleoside reverse transcriptase inhibitor-experienced patients: study TMC125-C227. HIV Med 2008; 9:883-96.
Steigbigel RT, Cooper DA, Teppler H, et al. Long-term efficacy and safety of Raltegravir combined with optimized background therapy in treatment-experienced patients with drug- resistant HIV infection: week 96 results of the BENCHMRK 1 and 2 Phase III trials. Clin Infect Dis 2010; 50:605-12.
Lazzarin A, Campbell T, Clotet B, et al. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-2: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet 2007; 370:39-48.
Madruga JV, Cahn P, Grinsztejn B, et al. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-1: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet 2007; 370:29-38.
Gulick RM, Lalezari J, Goodrich J, et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med 2008; 359:1429-41.
Yazdanpanah Y, Fagard C, Descamps D, et al. High rate of virologic suppression with raltegravir plus etravirine and darunavir/ritonavir among treatment-experienced patients infected with multidrug-resistant HIV: results of the ANRS 139 TRIO trial. Clin Infect Dis 2009; 49:1441-9.
Molina JM, Lamarca A, Andrade-Villanueva J, et al. Efficacy and safety of once daily elvitegravir versus twice daily raltegravir in treatment-experienced patients with HIV-1 receiving a ritonavir-boosted protease inhibitor: randomised, double-blind, phase 3, non- inferiority study. Lancet Infect Dis 2012; 12:27-35.
6. FACTORES QUE CONDICIONAN EL ÉXITO DEL TRATAMIENTO ANTIRRETROVIRAL
Adherencia
Se aconseja la lectura del Documento de consenso del Plan Nacional sobre el SIDA, GeSIDA y SEFH para mejorar la adherencia al TAR en pacientes con infección por VIH1.
En este documento se define la adherencia subóptima como primera causa de fracaso terapéutico y se abordan con detalle los factores que influyen en la adherencia al TAR, los métodos para evaluarla y las intervenciones para mejorarla. Entre las estrategias que han demostrado utilidad se encuentran el envío de mensajes recordatorios, la terapia cognitiva, las intervenciones educativas y de soporte, y el tratamiento directamente observado23. Las estrategias digitales basadas en internet o dispositivos móviles podrían ser eficaces para mejorar la adherencia1.
Un estudio reciente ha mostrado altas tasas de supresión virológica con estrategias de tratamiento intermitente basadas en INI administrados 4 o 5 días en semana en pacientes virológicamente suprimidos4. Por otro lado, en pacientes con supresión virológica se han observado cifras más elevadas de biomarcadores inflamatorios en aquellos con una adherencia inferior al 100%5. No está clara la implicación clínica de estos estudios, y con la evidencia clínica disponible no se recomiendan pautas de tratamiento intermitente.
La relación entre adherencia, control virológico y selección de resistencias varía entre las diferentes clases de FAR y la situación clínica del paciente (CVP, tiempo de viremia indetectable).En pacientes con adherencia subóptima la probabilidad de alcanzar viremia indetectable es mayor con pautas STR (single-tablet regimen) que con pautas MTR (multiple-tablet regimen)6.
Aún está por determinar la eficacia de los nuevos tratamientos inyectables ante una eventual adherencia subóptima al TAR. Sus potenciales ventajas incluyen la administración cada dos meses, la disminución del estigma y evitar la vía oral si hay dificultad para la deglución o la digestión. Sin embargo, no está claro cuál sería la mejor manera de manejar los abandonos o las dosis perdidas con estos tratamientos7. Una guía al respecto puede consultarse en la ficha técnica de los productos.
La polifarmacia se ha asociado a una menor probabilidad de supresión virológica8, y puede abordarse mediante intervenciones de desprescripción farmacológica. Se recomienda consultar el documento de GeSIDA sobre esta estrategia9.
La coformulación de FAR simplifica el TAR y previene la mala adherencia selectiva, mejorando la adherencia global10.
Recomendaciones
- Antes de iniciar el TAR se debe preparar al paciente, identificar y corregir las causas potenciales de adherencia incorrecta (A-III).
- Una vez iniciado el TAR se recomienda efectuar un contacto con el paciente a las 2‐4 semanas para comprobar la adherencia y corregirla si es preciso (A-III).
- La adherencia debe monitorizarse, reforzarse e intervenir activamente si fuese necesario coincidiendo con las visitas clínicas incluso en pacientes con CVP indetectable (A-III).
- El control de la adherencia debe realizarse idealmente por un equipo mulidisciplinar, adaptado a la disponibilidad de cada centro, que incluya médicos, personal de enfermería, profesionales de apoyo psicológico, farmacia hospitalaria y organizaciones comunitarias (A-III).
- En pacientes con adherencia subóptima se recomienda utilizar pautas de elevada barrera genética como las basadas en IP/p (de preferencia DRV) o en DTG o BIC para prevenir la selección de resistencias (A-II).
- Para conseguir una adherencia óptima, se recomienda que las pautas del TAR sean, en la medida de lo posible, de uno o dos comprimidos al día, en una única dosis de administración, y sin restricciones dietéticas, efectos secundarios ni interferencias con los hábitos de vida de los pacientes (A-II).
- En pacientes con evidencia o riesgo de mala adherencia selectiva se recomienda administrar pautas de antirretrovirales en comprimido único por ser la estrategia más eficiente para prevenirla (A-II).
Tolerabilidad y efectos adversos
6.2.1. FACTORES ASOCIADOS A LA TOLERABILIDAD DEL TAR
La tolerabilidad depende de aspectos relacionados con la toma de FAR (número y tamaño de los comprimidos, requisitos de administración, incidencia e intensidad de efectos secundarios inmediatos), pero también de factores dependientes del paciente (edad, sexo, peso, situación clínica y expectativas respecto al tratamiento). En el último decenio tanto los FAR como su posología han mejorado notablemente, particularmente con coformulaciones y regímenes de pastilla única, lo cual ha favorecido su aceptación por los pacientes1. A pesar de esta mejora, los efectos adversos de los FAR no son infrecuentes y pueden condicionar la adherencia y el éxito del TAR2. Las siguientes páginas web: https://hivinsite.ucsf.edu Guías actualizadas 11.0 https://www.eacsociety.org/media/final2021eacsguidelinesv11.0_oct2021.pdf , ofrecen información actualizada sobre los efectos adversos.
Los efectos adversos son una de las causas principales para cambiar un TAR que está siendo eficaz. En el capítulo 4 se comentan las toxicidades principales reales o potenciales del TAR, las evidencias para cambiarlo en un contexto de resolver un determinado efecto adverso o prevenir su aparición y las recomendaciones del panel de expertos de GeSIDA al respecto.
6.2.2. CLASIFICACIÓN CRONOLÓGICA DE LOS EFECTOS ADVERSOS
Los efectos adversos de los FAR pueden ser inmediatos o tardíos. Los efectos inmediatos se producen en los primeros días o semanas de tratamiento, mientras que los tardíos aparecen al cabo de meses o años después del inicio de éste. Se debe tener en cuenta que algunas circunstancias pueden favorecer la aparición de efectos adversos (ej. interacciones medicamentosas) y que los FAR también pueden exacerbar algunas patologías.
6.2.3. EFECTOS ADVERSOS INMEDIATOS
Los efectos adversos inmediatos están bien definidos. En algunos casos pueden anticiparse y suelen ser fáciles de controlar. Afectan principalmente a la esfera digestiva, cutánea o neuropsicológica, y su incidencia y factores asociados son generalmente conocidos.
Entre los FAR actualmente recomendados: los IP/p pueden producir efectos digestivos3; los ITINN de primera generación (particularmente NVP) hepáticos y RHS; ABC puede producir RHS en pacientes con HLA-B*5701 positivo, con un VPP del 50% y un VPN del 100%4; DRV exantema; y EFV, RPV, DOR y los cuatro INI actualmente disponibles (BIC, DTG, EVG y RAL) pueden producir efectos neuropsiquiátricos (Tabla 7). La experiencia post-comercialización sugiere que el insomnio o la discontinuación por efectos neuropsicológicos son inusuales con los INI disponibles, si bien son más frecuentes con DTG que con BIC, RAL o EVG567.
En los últimos años se ha autorizado el uso de FAR de administración parenteral con semividas prolongadas. Estos regímenes pueden facilitar la adherencia al TAR. En el caso de la pauta con CAB+RPV AP, el efecto adverso más frecuente (99% grado 1 o 2) fue el dolor en la zona de inyección (este efecto adverso disminuyó en las posteriores administraciones)89.
Recomendaciones
- La determinación del alelo HLA-B*5701 es obligada antes de prescribir ABC para evitar el riesgo de RHS(A-I).
- Se debe explicar al paciente cómo tomar correctamente una pauta de TAR y la posibilidad de que ocurran determinados efectos adversos inmediatos ya conocidos. Al iniciar un régimen de TAR se debe explicar qué actitud debe tomar el paciente si ocurre un determinado efecto adverso y, en cualquier caso, se debe facilitar siempre la posibilidad de comunicación directa con el médico (A-III).
- Se debe efectuar, en cada visita de control, un interrogatorio exhaustivo acerca de la presencia o ausencia de los potenciales efectos adversos del TAR que esté recibiendo el paciente (A-III).
- Los efectos adversos inmediatos leves se pueden tratar sintomáticamente, valorando la evolución de la tolerabilidad del paciente (A-III); si el efecto adverso tiene gran intensidad o duración prolongada o no es asumible por el paciente, se debe cambiar el o los FAR potencialmente implicados (A-I).
6.2.4. EFECTOS ADVERSOS TARDÍOS
Los efectos adversos tardíos se conocen menos que los inmediatos y son más difíciles de prever y controlar; potencian los síntomas de las enfermedades crónicas asociadas al envejecimiento y afectan al funcionamiento de órganos y sistemas.
El perfil de órganos y sistemas a los que pueden afectar los diversos FAR, así como los factores de riesgo asociados a tal afectación, no se conoce en su totalidad, particularmente en lo que concierne a los FAR más recientes. La Tabla 8 resume los efectos secundarios tardíos más característicos de los FAR actuales3.
El papel que suelen tener los FAR en la producción o desarrollo de enfermedades crónicas es en general pequeño y mucho menor que el de otros factores de riesgo clásicos ya conocidos en la población general que, en algunos casos, están sobrerrepresentados en pacientes con infección por el VIH, como son el consumo de tabaco, alcohol u otras drogas, una dieta inadecuada, o la ausencia de ejercicio físico.
El riesgo absoluto de efectos secundarios tardíos con los FAR actualmente recomendados es pequeño y no cuestiona el beneficio global del inicio del TAR. Sin embargo, en pacientes de alto riesgo o con enfermedades crónicas ya diagnosticadas, determinados FAR pueden contribuir, desencadenar o desarrollar tales enfermedades.
La retirada de algunos FAR implicados en efectos adversos tardíos puede mejorar, al menos parcialmente, la alteración subyacente, aunque se desconoce si tal modificación puede alterar la historia natural de la enfermedad crónica en cuestión o la supervivencia. Los FAR contribuyen de forma colateral al riesgo o progresión de determinadas enfermedades crónicas, pero hay otros factores generalmente más importantes, cuyo abordaje suele ser prioritario.
Señalar, en relación con los INI, que cada vez hay más estudios observacionales que sugieren que su uso podrían conducir a aumentos estadísticamente significativos del peso corporal e incluso de obesidad10. Este efecto, que se observa con más frecuencia en mujeres y pacientes de raza negra, se ha descrito especialmente con el uso de DTG y BIC, y podría estar en parte agravado por el uso concomitante de TAF11. Con la pauta de CAB+RPV AP se ha descrito, en la semana 48, una mediana de aumento de peso de +1,80 kg en el grupo de terapia de acción prolongada y de +0,30 kg en el de vía oral1213.
Como con otros fármacos, por el momento se desconocen las causas de la ganancia de peso. En cuanto a los posibles efectos adversos a largo plazo de CAB+RPV AP la observación más allá de 96 semanas en los ensayos clínicos no ha mostrado hasta ahora un perfil de seguridad diferente a las pautas orales con compuestos similares89.
El manejo del impacto de determinados antirretrovirales sobre los efectos secundarios crónicos puede contemplar tanto la retirada del fármaco en cuestión como el tratamiento de la condición crónica de forma similar a la recomendada en la población general. De estas dos estrategias, es más eficaz el tratamiento de la condición crónica1415 aunque pueden ser necesarias ambas.
Recomendaciones
- Se debe individualizar el TAR evaluando el riesgo y/o la presencia de enfermedades crónicas de manera que la pauta elegida no contenga FAR que puedan favorecer la aparición o progresión de las mismas (A-II).
- Se debe efectuar, en cada visita de control, un interrogatorio exhaustivo acerca de la presencia o ausencia de los potenciales efectos adversos del TAR que esté recibiendo el paciente (C-III).
- En los pacientes tratados con INI, y en especial con la asociación de DTG o BIC y TAF, se recomienda monitorizar el peso y el riesgo cardiovascular (A-I).
Interacciones farmacológicas
El mayor empleo de INI no potenciados en la actualidad ha disminuido globalmente envejecimiento, y la mayor presencia de comorbilidades y polifarmacia en la población con VIH, las pDDI siguen teniendo una elevada prevalencia1234567. La presencia de pDDI se asocia con un mayor coste sanitario4. Entre las co-medicaciones más frecuentemente implicadas con pDDI con FAR se encuentran los corticoides inhalados, quetiapina, antiagregantes, inhibidores de la bomba de protones y los cationes polivalentes. También es frecuente el consumo de productos de herboristería, suplementos dietéticos, complementos para ganar masa muscular, medicinas alternativas o drogas recreativas asociadas al sexo (chemsex)5 que pueden presentar pDDI graves.
En grandes estudios de cohortes se ha observado una prevalencia de pDDI con FAR de hasta el 45%13, considerándose graves entre un 2 y 5% de ellas13. En mayores de 65 años esta proporción puede ser del 16%4. Un mayor riesgo de pDDI graves se asoció con polifarmacia (asociación de 5 o más fármacos)12, edad1, coinfección por VHC3 y con el uso de IP/p, EVG/c o ITINN(1-4,6). Por el contrario, un menor riesgo se asoció con el uso de INI no potenciados(1-4,6), especialmente cuando no se utilizaban con TDF6. BIC/FTC/TAF por sus características metabólicas puede presentar un moderado mayor riesgo de pDDI que pautas basadas en DTG o RAL6, aunque en todo caso las asociaciones contraindicadas con BIC son infrecuentes (en <0,25% de los pacientes)7 y asociadas mayoritariamente al uso concomitante de inductores enzimáticos potentes.
De los estudios anteriores se deduce que las interacciones siguen teniendo un papel importante, especialmente en pacientes mayores polimedicados. Consultar el documento https://www.mscbs.gob.es/ciudadanos/enfLesiones/enfTransmisibles/sida/ documentos/110521_Doc_CONSENSO_ENVEJECIMIENTO_Y_VIH.pdf. (actualización 2021, interacciones en apartados 8.3 y 9.5.5).
Existen diversas páginas web que permiten consultar pDDI:
http://www.hiv-druginteractions.org (en inglés y español)
http://app.hivclinic.ca (en inglés)
http://www.hivmedicationguide.com (en inglés y francés)
http://www.interaccionesvih.com (en español)
http://www.clinicalcasesddis.com/ (en inglés) Esta nueva web permite consultar casos reales de interacciones y notificar casos propios. Puede ser muy útil cuando la información es escasa.
App: InterApp ARV (en español)
El artículo de Nhean S et al.8 revisa este tema en profundidad.
La información científica se renueva constantemente por las compañías farmacéuticas y las autoridades sanitarias: Agencia Europea del Medicamento-EMA (https://www.ema. europa.eu/en/medicines) y FDA (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/). En la parte III de las guías EACS están disponibles tablas de consulta sobre pDDI.
En la Tabla 9 de estas guías se indican las pDDI de mayor gravedad (contraindicadas o no recomendadas) de los FAR más empleados actualmente, así como de los más nuevos.
Globalmente, las interacciones más relevantes son las farmacocinéticas (dan lugar a una modificación de concentraciones), en especial las que afectan al metabolismo. El citocromo P450 (CYP) es la vía metabólica más importante y su principal isoenzima es CYP3A4. Algunos FAR son metabolizados por esta vía y pueden ser víctima de los fármacos inductores, que pueden reducir su eficacia.
Los principales inductores potentes del CYP3A4 son: apalutamida, carbamazepina, enzalutamida, fenitoína, fenobarbital, lumacaftor, mitotano, primidona, rifampicina e Hypericum.
Los principales inductores moderados del CYP3A4 son: bexaroteno (>300 mg/m2/ día), bosentan, cenobamato, dabrafenib, efavirenz, eslicarbazepina (débil-moderado), etravirina, lorlatinib, modafinilo, midosturina, rifabutina y rifapentina.
Otra vía metabólica es la conjugación de los FAR o de sus metabolitos con otros productos, por ejemplo, la glucuronidación (UDPGT). Inductores potentes de la glucuronidación: rifampicina. Existe menos información con otros como carbamazepina, fenitoína, fenobarbital y primidona.
Los FAR son sustratos de uno o varios sistemas enzimáticos y a la vez pueden comportarse como inductores y/o inhibidores de los mismos. La inducción producirá una disminución de las concentraciones del otro fármaco (sustrato), pudiendo disminuir su eficacia. La inhibición ocasionará un aumento de las concentraciones, lo que supone un mayor riesgo de toxicidad. En general, la inducción es un proceso lento (días o semanas), mientras que la inhibición se produce rápidamente (horas). Algunos FAR pueden ser inhibidores e inductores al mismo tiempo, predominando uno u otro efecto.
Los FAR que presentan un mayor número de interacciones, que pueden ser potencialmente graves, son los IP/p y EVG/c (inhibidores potentes del metabolismo) y los ITINN (EFV, ETR y, con menor intensidad, NVP, inductores del metabolismo). Al evaluar las interacciones debemos considerar la capacidad de cada fármaco (tanto los FAR como los concomitantes) de actuar como causantes o como receptores/víctimas de éstas.
Los INI no potenciados, los ITINN RPV y DOR, los ITIAN e ibalizumab tienen un perfil de interacciones más favorable porque no son inductores ni inhibidores enzimáticos (no son causantes de interacciones). Algunos de estos fármacos pueden ser útiles en pacientes polimedicados para evitar riesgos. Sin embargo, tanto INI no potenciados como RPV y DOR son, en mayor o menor medida, metabolizados mediante el CYP3A4 o UDPGT. Por este motivo pueden ser víctimas de interacción (especialmente con inductores potentes). Debemos tener también presentes las interacciones de los antiácidos y otros cationes con INI (incluyendo CAB durante la administración oral), RPV y ATV, y de los inhibidores de la bomba de protones con RPV y ATV, que pueden reducir la absorción de estos FAR.
CAB y RPV son los primeros FAR de acción prolongada que se administran por vía IM. Deben administrarse en la zona ventroglútea (recomendada) o dorsoglútea. Se están investigando otros lugares de inyección como el muslo. En pacientes anticoagulados la experiencia es muy limitada ya que en los ensayos clínicos se excluyeron estos pacientes. Por ello no se puede establecer recomendaciones específicas más allá de un uso con precaución y valoración del riesgo/beneficio. CAB es metabolizado por glucuronidación y carece de efecto inductor o inhibidor sobre el metabolismo. Es inhibidor del transportador OAT1/3 y se recomienda precaución con metotrexate. Además de la mayor comodidad en su posología, tienen la ventaja de obviar las interacciones que afectan a la absorción intestinal (como los inhibidores la bomba de protones o cationes polivalentes) y la necesidad de administrar RPV con alimentos. Sin embargo, persisten las interacciones que afectan al metabolismo. Con RPV IM, a diferencia de la oral, no es posible ajustar la dosis en presencia de inductores moderados del CYP3A4. Es por ello que CAB+RPV IM está contraindicado en presencia de inductores moderados o potentes del CYP3A4. Respecto a los inhibidores, tanto los inhibidores potentes del CYP3A4 como los inhibidores potentes de la glucuronidación se pueden emplear sin necesidad de ajuste de dosis. RPV puede prolongar el intervalo QT a dosis superiores a las terapéuticas y la FT recomienda precaución con fármacos que puedan aumentar sus concentraciones. Sin embargo, hasta el momento no se ha descrito Torsade de pointes, según la web https://www.crediblemeds.org/. La concentración plasmática máxima de RPV IM (600 mg c/mes o 900 mg c/2 meses) es similar a la observada con 25 mg/día vía oral. Hodge D et al.9 revisan las características farmacocinéticas y perfil de interacciones de esta nueva combinación de TAR.
Con respecto a los nuevos FAR en terapia de rescate ibalizumab, por ser un anticuerpo monoclonal, no se espera que presente interacciones farmacocinéticas. Fostemsavir, es un profármaco de temsavir. Se metaboliza principalmente por las esterasas plasmáticas y el CYP3A4 es minoritario. Por lo tanto, tiene poco riesgo de ser víctima de interacciones. Emplea los transportadores P-gp y BCRP. Está contraindicado con inductores potentes del CYP3A4 por riesgo de fracaso. En cambio, sí puede administrarse con inductores moderados como rifabutina. También puede combinarse con inhibidores del CYP3A4. Fostemsavir como causante de interacciones es inhibidor de algunos transportadores (OATP1B1 y BCRP). A tener en cuenta que todas las estatinas (excepto pravastatina) emplean el OATP para su eliminación biliar. Fostemsavir aumenta las concentraciones de etinilestradiol, por lo que se recomienda limitar la dosis de etinilestradiol a 30 mcg al día. Además prolonga el intervalo QT a dosis 4 veces superiores a las terapéuticas, por lo que el riesgo es mayor en presencia de inhibidores del CYP3A4.
Ciertos transportadores, como la glicoproteína-P (P-gp), OATP, OCT2 y MATE1, entre otros, pueden alterar la biodisponibilidad de algunos FAR y su distribución por el organismo o verse afectados por algunos FAR, como se ha descrito previamente. Por ejemplo, metformina es sustrato del transportador OCT2. DTG y, en menor medida, BIC son inhibidores de OCT2 10. Por ello, en asociación con DTG debe considerarse el ajuste de dosis de metformina, especialmente en pacientes con función renal alterada.
En cambio, con BIC no se recomienda modificar la dosis de metformina en pacientes con función renal normal. En pacientes con insuficiencia renal moderada, se debe considerar una monitorización estrecha al comienzo de la administración concomitante y considerar un ajuste de la dosis de metformina si fuera necesario. Puede encontrarse información sobre transportadores en la web http://dbts.ucsf.edu/fdatransportal.
Algunas interacciones farmacodinámicas (modificación del efecto, sin cambios en las concentraciones) tienen interés clínico. Algunos ejemplos son el incremento de toxicidad renal al asociar fármacos nefrotóxicos con TDF o la asociación de medicamentos que pueden prolongar el QTc.
Se han descrito interacciones importantes para el tratamiento del VHC y los FAR, que pueden consultarse en páginas web específicas http://app.hivclinic.ca, http:// www.hivmedicationguide.com/, http://www.hep-druginteractions.org/, así como en las recomendaciones elaboradas por la Agencia Española del Medicamento y Productos Sanitarios.
Las estrategias multidisciplinares de control de fármacos que involucran a profesionales de medicina, enfermería y farmacia11 o las aplicaciones para dispositivos móviles pueden ser herramientas útiles para disminuir el riesgo de interacciones en pacientes con el VIH tratados con FAR.
Recomendaciones:
- Se debe reseñar en la historia clínica todos los medicamentos, productos naturales, medicinas alternativas, suplementos dietéticos, complementos para ganar masa muscular o drogas recreativas que toma el paciente para evaluar posibles interacciones entre ellos (A-III).
- Ante cualquier riesgo de interacción se recomienda consultar las fichas técnicas y la información actualizada por las compañías farmacéuticas y las autoridades sanitarias (AIII). Se deben tener en cuenta las contraindicaciones y realizar ajustes de dosis cuando sea necesario (A-I).
- Cuando existe riesgo de interacciones farmacocinéticas entre dos o más fármacos, no se dispone de información específica y existe la posibilidad de determinar niveles plasmáticos, debe considerarse la monitorización para evitar toxicidad o ineficacia terapéutica (B-III).
- En pacientes mayores con VIH se recomienda tener en cuenta los criterios de prescripción adecuada en geriatría consultado los documentos específicos (BIII).
- Considerar el uso de DTG o RAL como núcleo del TAR si el riesgo de interacciones es elevado, desconocido o incontrolable (A-II). Alternativamente, si se descarta el uso de inductores enzimáticos potentes (ver texto) se puede considerar BIC (B-II) o DOR , esta última con menos evidencia (C-II).
- CAB+RPV AP es una opción recomendable siempre que no se asocie a inductores enzimáticos moderados o potentes de las enzimas CYP3A (B-III).
Tablas:
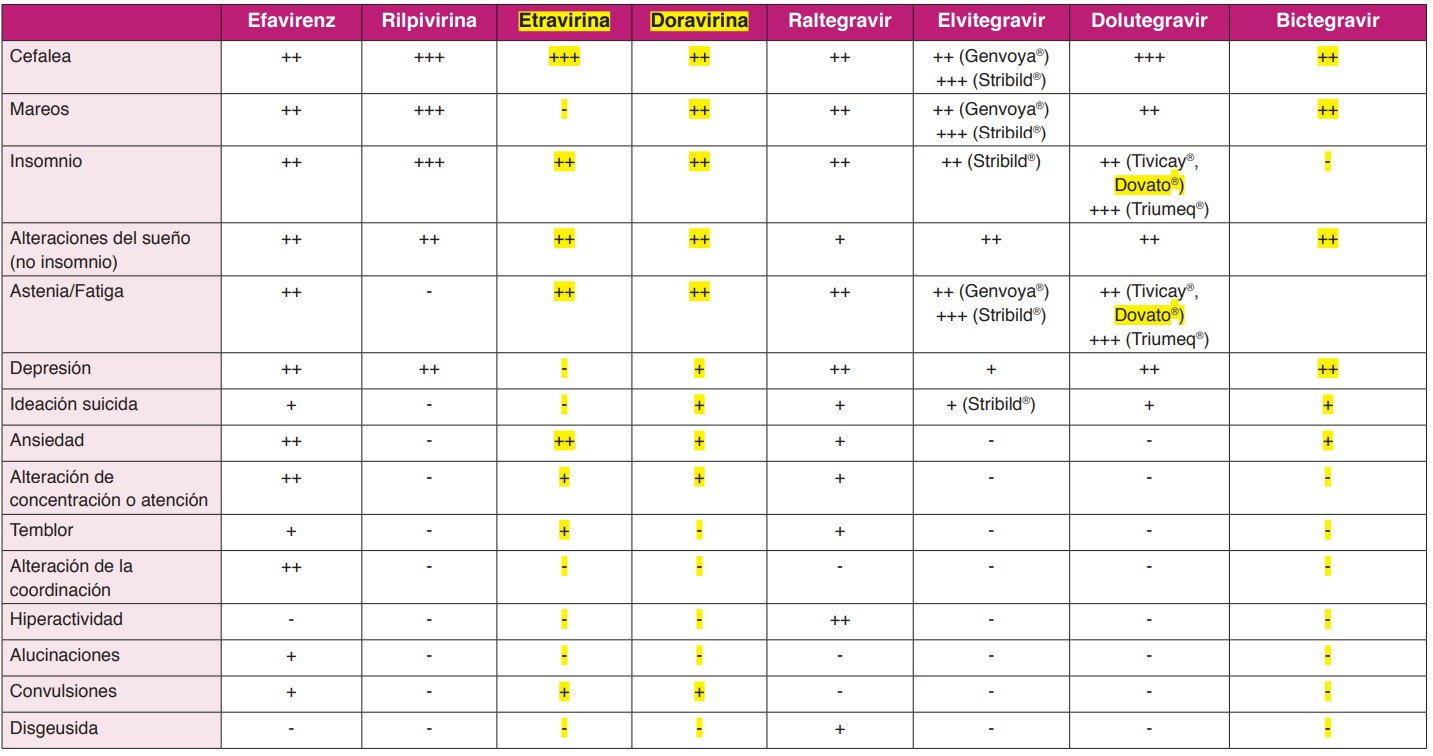
*Según la ficha técnica más reciente disponible en www.ema.europa.eu. Frecuencia de efectos adversos: +++, >10%; ++, 1-10%; +, 0.1-1%
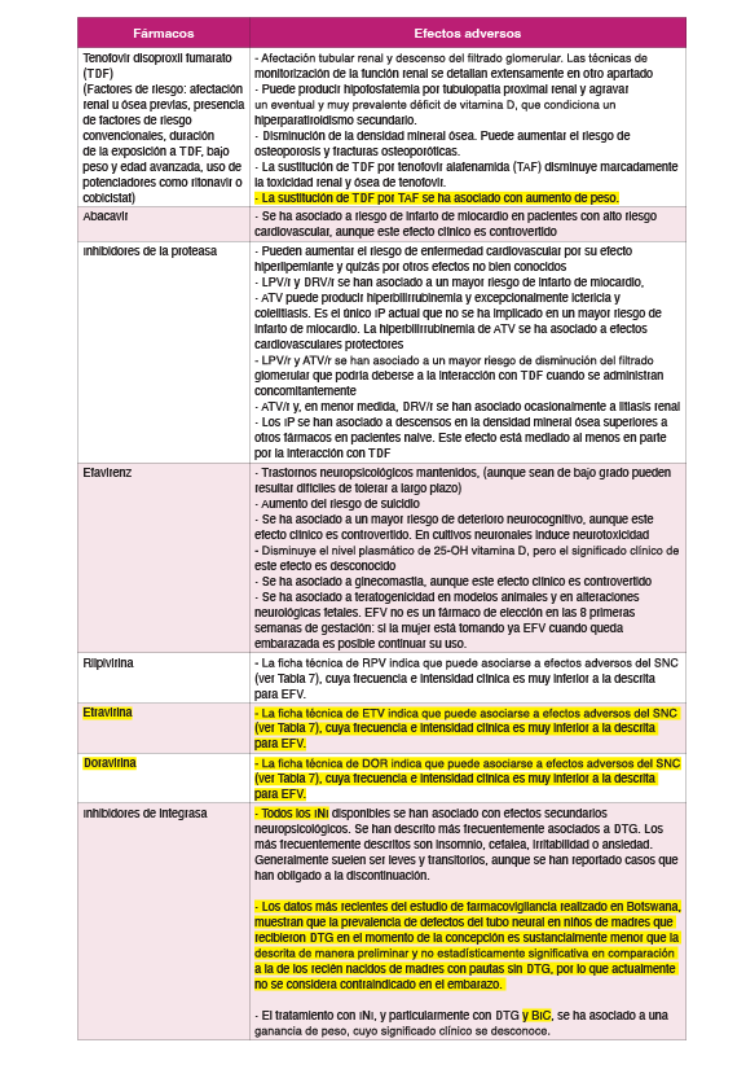
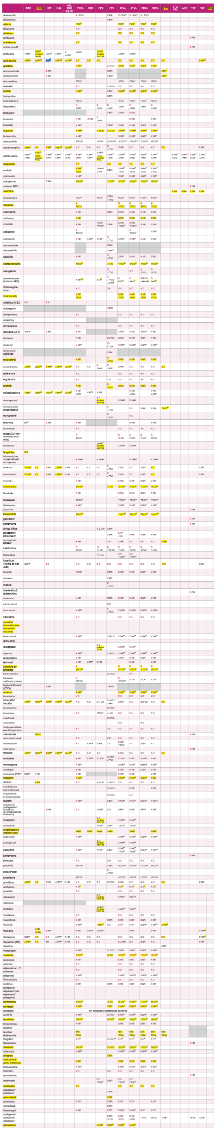
Nota: no se han tenido en cuenta los siguientes fármacos por no estar disponibles en nuestro país (ni mediante importación como medicamento extranjero) o estar en desuso: bepridil, boceprevir, cisaprida, dextropropoxifeno, flibanserina, fosamprenavir, indinavir, levometadil, lurasidona, rolapitant, simeprevir, terfenadina y tipranavir. Algunos de ellos fueron retirados del mercado por toxicidad. A pesar de no estar incluidos en la tabla, podrían tener contraindicaciones con algunos antirretrovirales.
Abreviaturas:
C: asociaciones “contraindicadas” o “no debe emplearse” según la ficha técnica europea.
NR: asociaciones “no recomendadas” según la ficha técnica europea.
TOX: no asociar por riesgo de aumento de toxicidad, ya sea por interacción farmacodinámica o farmacocinética.
FCO: no asociar por reducción de niveles plasmáticos/eficacia del fármaco.
ARV: no asociar por disminución de niveles plasmáticos/eficacia del antirretroviral.
ARV: no asociar por aumento de niveles plasmáticos/toxicidad del antirretroviral.
¿?: dada la incertidumbre, es preferible emplear otras combinaciones, ya sea sustituyendo el antirretroviral o el fármaco asociado, en función de las posibilidades.
Superíndices:
1 Si no es posible evitar la coadministración, se debe reducir la dosis de abemaciclib: en pacientes con 150mg/12h reducir a 100 mg dos veces al día; en pacientes con dosis reducida de 100 mg dos veces al día reducir a 50 mg dos veces al día; en pacientes con dosis reducida de 50 mg dos veces al día mantener la dosis de abemaciclib con un estrecho control de los signos de toxicidad. Alternativamente, se puede reducir la dosis a 50 mg una vez al día o suspender el tratamiento. En caso de suspender el inhibidor de CYP3A4, la dosis de abemaciclib se debe aumentar a la dosis utilizada previamente (después de 3 a 5 semividas del inhibidor CYP3A4).
2 Antiácidos: En general, presencia de resistencia a INI, evitar cualquier antiácido en combinación con INI.
- BIC: con Al o Mg: BIC 2h antes del antiácido o junto con alimentos 2 horas después del antiácido. No se recomienda la administración concomitante con sucralfato. Con Fe: BIC al menos 2 horas antes del suplementos de hierro, o tomar los dos juntos con
- CAB oral: administrar CAB 4 horas antes o 2 horas después del antiácido.
- DTG: debe tomarse como mínimo 2h antes o 6h después de los preparados que contengan Al (incluyendo sucralfato), Mg o multivitamínicos. Con carbonato de calcio o fumarato ferroso puede administrarse el dolutegravir conjuntamente con el antiácido siempre que se tomen con
- EVG/cobi: se recomienda espaciarlo 4h de los antiácidos.
- RAL QD: evitar antiácidos ya sea juntos o separados, incluso los de
- RAL BID: con Al (incluyendo sucralfato) y Mg probablemente sea mejor evitar la asociación (la ficha técnica europea no recomienda la administración simultánea y la americana ni la simultánea ni espaciados). Con Ca: interacción sin importancia clínica.
- ATV: administrar ATV 2h antes de los antiácidos ó 1h después de los
- RPV: administrar RPV 4h antes ó 2h después de los antiácidos.
3 Edoxaban podría emplearse a mitad de dosis 30 mg/día. Dabigatran administrado de forma simultánea con ritonavir podría suponer una alternativa más segura que rivaroxaban, aunque los datos son escasos. Se han publicado dos casos de asociación de dabigatran con ATV/r y LPV/r sin interacción significativa. En cambio, el uso de dabigatran con cobicistat podría aumentar el riesgo de sangrado. Probablemente sea porque cobicistat es más inhibidor de la P-gp intestinal que ritonavir. Con apixaban, la ficha técnica americana recomienda para una dosis equivalente a 5 mg/12h, al añadir un inhibidor potente del CYP3A4 y P-gp (como ritonavir o cobicistat), ajustar dosis de mantenimiento a 2,5 mg/12h, mientras que si la dosis inicial es de 2,5 mg/12h, evitar la asociación. Una serie de 6 casos apoya este ajuste (la dosis inicial empleada en TVP fue de 5 -10 mg/12h los 7 primeros días, seguido de 2,5 mg/12h). Sin embargo, la ficha técnica europea no recomienda apixaban con inhibidores potentes de CYP3A4+P-gp.
4 También atazanavir no potenciado.
5 EFV: si se requiere coadministración de ATV con un ITINN como EFV, en pacientes naïve podría considerarse ATV/r 400/100 mg c/24h con cuidadosa monitorización. En pacientes pretratados no se recomienda. No se puede emplear ATV no potenciado ni ATV potenciado con cobicistat.
6 DOR asociada a inductores moderados del CYP3A4: con rifabutina: aumentar DOR a 100 mg dos veces al día (espaciados aproximadamente 12 horas). Con el resto de inductores moderados no se ha estudiado. Si no es posible evitar la combinación, aumentar DOR a 100 mg dos veces al día (espaciados aproximadamente 12 horas).
7 Si no es posible evitar la coadministración con un inhibidor potente del CYP3A con bosutinib, reducir la dosis de bosutinib o interrumpir el tratamiento.
8 Corticoides metabolizados mediante el CYP3A4: sus concentraciones pueden aumentar en presencia de inhibidores de esta enzima como ritonavir o cobicistat. La ficha técnica de cobicistat a nivel teórico no recomienda ninguno de ellos. La ficha técnica de ritonavir no recomienda fluticasona, budesonida, ni triamcinolona y recomienda precaución con dexametasona y prednisolona/prednisona. Dexametasona puede emplearse en dosis únicas pero en dosis múltiples se ha descrito sd. Cushing incluso con gotas oftálmicas junto con IP potenciados con RTV. En caso de que deban emplearse estas asociaciones, se recomienda una monitorización cuidadosa de los efectos terapéuticos y las reacciones adversas. Debe considerarse el uso de corticoides alternativos como beclometasona. En caso de retirada del glucocorticoide, se puede requerir una reducción progresiva de la dosis durante un mayor periodo de tiempo.
9 DTG: en pacientes sin resistencia a INI, aumentar DTG a 50 mg c/12 h hasta 2 semanas después de finalizar rifampicina y en pacientes con resistencia a INI considerar otras alternativas terapéuticas.
10 El carbón activo puede retener fármacos en su superficie dificultando su absorción. La información es escasa y se desconoce el tiempo que deben separarse los fármacos para evitar la interacción. Evitar en lo posible.
11 Ceritinib, mediante la inhibición del CYP3A4, puede aumentar los niveles de RPV. Ambos fármacos pueden prolongar el intervalo QT, por lo que se recomienda precaución.
12 Si no es posible evitar la coadministración, reducir la dosis de ceritinib a un tercio aproximadamente (dosis no comprobada clínicamente), redondeándola al múltiplo de 150 mg más cercano. Si el paciente tolera bien una dosis reducida, ésta puede incrementarse de nuevo vigilando estrechamente la seguridad, para evitar que el paciente resulte insuficientemente tratado. Después de la interrupción del tratamiento con el inhibidor potente de CYP3A, restablecer la dosis previa.
13 Si insuficiencia renal: ClCr 30-60 mL/min: reducir 50% la dosis de claritromicina; ClCr<30 mL/min: reducir 75% la dosis de claritromicina.
14 Una alerta de la AEMPS desaconseja el uso de colchicina junto con inhibidores de CYP3A4 o de la glicoproteína-P.
15 La administración conjunta con fármacos que prolongan el intervalo QT y/o inducen Torsade de Pointes está contraindicada.
16 La administración conjunta con fármacos que prolongan el intervalo QT no está recomendada (en caso imprescindible, monitorizar electrocardiograma)
17 Precaución por riesgo de prolongar el intervalo QT.
18 EFV: junto con DRV/r 800/100 mg c/24h puede dar lugar a una Cmín de DRV subóptima. Debe emplearse DRV/r 600/100 mg c/12h.
19 Antes de iniciar eliglustat se debe genotipar el CYP2D6. En metabolizadores ultrarrápidos del CYP2D6 no debe emplearse eliglustat en ninguna circunstancia. El uso de inhibidores potentes o moderados del CYP3A4 combinado con inhibidores potentes o moderados del CYP2D6 está contraindicado. RTV y COBI son inhibidores potentes del CYP3A4 y débiles del CYP2D6: su uso está contraindicado en metabolizadores lentos del CYP2D6 y debe emplearse con precaución en metabolizadores intermedios o rápidos del CYP2D6. Sería preferible emplear TAR con INI no potenciados. En caso imprescindible, consultar la ficha técnica del fármaco (ajustes de dosis complejos basados en la función renal, función hepática e interacciones).
20 IBP: ATV/c: no se recomienda. ATV/r: no se recomienda. Si la combinación de ATV/r con un IBP es inevitable, valorar ATV/r 400/100 mg c/24h y no exceder una dosis de omeprazol de 20 mg c/24h (o dosis equivalente de otro IBP).
21 DTG: en pacientes con resistencia a INI no se debe utilizar con ETR sin la administración concomitante de ATV/r, DRV/r o LPV/r (recordar que ETR no debe asociarse a cobicistat.
22 antagonistas H2:
ATV/c: para los pacientes que NO tomen tenofovir DF, se debe administrar ATV/c una vez al día con alimentos simultáneamente con o por lo menos 10 horas después de una dosis del antagonista del receptor-H2. La dosis del antagonista del receptor-H2 no debe exceder una dosis comparable a 20 mg de famotidina dos veces al día. Para los pacientes que SI tomen tenofovir DF, no se recomienda administrar de forma conjunta ATV/c con un antagonista del receptor-H2.
ATV/r-antagonistas H2: para pacientes que NO estén tomando tenofovir DF, si se administra de forma conjunta ATV/r 300/100 mg y antagonistas de receptores-H2, no se debe superar una dosis equivalente a 20 mg de famotidina dos veces al día. Si fuese necesaria una dosis mayor de un antagonista de receptor-H2 (p. ej. 40 mg de famotidina dos veces al día o equivalente) se puede considerar un aumento de la dosis de ATV/r desde 300/100 mg hasta 400/100 mg. Para pacientes que SI tomen tenofovir DF, si se administra de forma conjunta ATV/r y un antagonista de receptor-H2, se recomienda un aumento de ATV/a 400/100 mg. No se debe superar una dosis equivalente a 40 mg de famotidina dos veces al día.
RPV-antagonistas H2: solamente se deben utilizar los antagonistas de los receptores H2 que se puedan administrar una vez al día. Éstos deben administrarse al menos 12 horas antes o al menos 4 horas después de rilpivirina.
23 Información escasa. Valorar riesgo/beneficio. Si es posible, emplear otras combinaciones.
24 Tacrolimus + ritonavir: interacción muy importante, puede requerir dosificación una vez por semana. Tacrolimus + cobicistat datos limitados indican que la interacción es algo menor que con ritonavir, puede requerir dosificación c/3-4 días. Ajustar según concentraciones plasmáticas.
25 La administración concomitante de glecaprevir/pibrentasvir con ATV/r está contraindicada debido al riesgo de elevaciones de la ALT. Se produce un aumento importante de los niveles de glecaprevir (AUC 6 veces mayor) y, en menor medida, de pibrentasvir.
26 itraconazol/ketoconazol: no se recomiendan dosis del antifúngico mayores a 200 mg c/24h.
27 Basándose en estudios farmacocinéticos, si el uso de lapatinib con inhibidores potentes del CYP3A4 no puede evitarse, podría valorarse una reducción de dosis de 1250 mg a 500 mg al día para alcanzar niveles similares a los obtenidos con la dosis habitual de lapatinib en ausencia de estos inhibidores. Cuando el inhibidor potente se suspende, una semana después debe volver a aumentarse la dosis de lapatinib a 1250 mg al día.
28 EFV: junto con LPV/r se recomienda aumentar la dosis de LPV/r a 600/150 mg c/12h.
29 La ficha técnica de rilpivirina recomienda aumentar la dosis a 50 mg/día en presencia de rifabutina, basado es estudios farmacocinéticos. Dado que no hay experiencia clínica con este ajuste de dosis, se recomienda valorar otras pautas de TAR.
30 Si no es posible evitar la coadministración, se recomienda reducir la dosis de olaparib a 100 mg dos veces al día (equivalente a una dosis total diaria de 200 mg).
31 Durante el tratamiento con ombitasvir/paritaprevir/ritonavir/dasabuvir puede administrarse ATV ó DRV suspendiendo temporalmente la potenciación con ritonavir, dado que el fármaco para el tratamiento del VHC ya va potenciado con ritonavir.
32 Si la administración concomitante es inevitable, se debe reducir la dosis de palbociclib a 75 mg al día. Si se interrumpe la administración del inhibidor potente, se debe aumentar la dosis de palbociclib (tras 3-5 semividas del inhibidor: aprox 36-60h para los IP) hasta la dosis utilizada antes de comenzar la administración del inhibidor potente del CYP3A.
33 Se recomienda evitar la administración concomitante y si no es posible, reducir la dosis de pazopanib a 400mg/día y monitorizar las reacciones adversas y reducir la dosis de nuevo si se observan reacciones adversas relacionadas con el fármaco.
34 Según ficha técnica, cuando se administre ponatinib con inhibidores potentes del CYP3A, se recomienda reducir la dosis inicial de ponatinib a 30 mg.
35 Según ficha técnica, evitar el uso de ribociclib con los inhibidores potentes de CYP3A4 por riesgo de prolongación del intervalo QT clínicamente significativa. Si no se puede evitar, reducir la dosis a 400 mg/24h. Sin embargo, no hay datos clínicos con este ajuste y debido a la variabilidad interpaciente, se recomienda un seguimiento estrecho. En caso de toxicidad, reducir dosis o interrumpir el tratamiento temporalmente. Si se suspende el inhibidor potente de CYP3A4, después de al menos 5 semividas del inhibidor de CYP3A4, restablecer la dosis previa de ribociclib.
36 silodosina: la ficha técnica de silodosina no recomienda el uso concomitante de inhibidores potentes del CYP3A4 (aumento del riesgo de hipotensión ortostática). En caso imprescindible, en pacientes que reciban ritonavir o cobicistat podría valorarse el uso de dosis bajas de tamsulosina 0,4 mg/día o silodosina (empezar con 4 mg al día). Como alfabloqueantes no uroselectivos podrían emplearse terazosina (que se metaboliza mínimamente en el hígado) u otros con mayor metabolismo hepático que deberán iniciarse dosis bajas y con estrecha monitorización, como doxazoxina ó prazosina. El inhibidor de la 5-alfa-reductasa finasterida es metabolizado mediante el YP3A4 pero tiene amplio margen terapéutico.
37 Evitar el uso de inhibidores potentes del CYP3A4 con sunitinib (aumento del riesgo de prolongación del intervalo QT). Si esto no es posible, puede que sea necesario reducir las dosis de sunitinib hasta un mínimo de 37,5 mg al día en el caso de tumor del estroma gastrointestinal (GIST) y carcinoma de células renales metastásico (CCRM) o de 25 mg al día para tumores neuroendocrinos pancreáticos (pNET), junto con una cuidadosa monitorización de la tolerabilidad.
38 Los inhibidores del CYP3A4 como ritonavir o cobicistat pueden reducir la formación del metabolito activo de clopidogrel, y, como consecuencia, la eficacia del tratamiento antiagregante, poniendo al paciente en riesgo de recurrencia de su enfermedad coronaria. Efavirenz también puede alterar la formación del metabolito activo de clopidogrel y se ha descrito un caso de trombosis del stent. Prasugrel supone una alternativa más segura desde el punto de vista de las interacciones. A pesar que es metabolizado mediante el CYP3A4, ni inductores potentes ni inhibidores potentes modificaron su eficacia antiagregante plaquetaria. No se recomienda el uso de prasugrel en edad ≥75 años, por aumento del riesgo de hemorragia. Ticagrelor contraindicado en presencia de inhibidores del CYP 3A4 por aumento del riesgo de hemorragia; en cambio, los inductores pueden reducir su eficacia.
39 tolterodina prolonga el intervalo QTc. La ruta metabólica principal está mediada por el enzima polimórfico CYP2D6, formando un metabolito 5-hidroximetílico con igual actividad al fármaco de origen. Un 7% de la población son metabolizadores lentos y en ellos las concentraciones plasmáticas son unas 7 veces mayores y el CYP3A4 pasa a ocupar un papel importante en el metabolismo. Si se añade un fármaco que es inhibidor potente del CYP3A4 como ritonavir o cobicistat las concentraciones de tolterodina podrían ser todavía mayores y aumentar el riesgo de prolongación del QT.
40 Según datos de ficha técnica, el uso concomitante de venetoclax con inhibidores potentes del CYP3A4, está contraindicado al inicio y durante la fase de ajuste de la dosis debido al mayor riesgo de síndrome de lisis tumoral. Durante esta fase debería valorarse un cambio de TAR como por ejemplo a raltegravir, dolutegravir o bictegravir. Para los pacientes que hayan completado la fase de ajuste de dosis y estén recibiendo una dosis diaria constante de venetoclax, si el uso de ritonavir/cobicistat es imprescindible, la dosis de venetoclax debe reducirse a un 25% (reducción del 75%). Se recomienda monitorización estrecha de efectos adversos y ajustar la dosis si es necesario. En caso de cambio de TAR a otro que no sea inhibidor potente del CYP3A4, deberá restablecerse la dosis estándar de venetoclax a los 2 ó 3 días después de la interrupción del IP o cobicistat.
41 voriconazol: no debe emplearse a menos que una evaluación riesgo-beneficio justifique el uso. Se debe determinar, si es posible, el genotipo CYP2C19 de los pacientes. Si la combinación es inevitable, las recomendaciones según el estatus del CYP2C19 son las siguientes: a) en pacientes con al menos un alelo CYP2C19 funcional, se recomienda estrecha monitorización clínica por la pérdida de eficacia de ambos fármacos. b) en pacientes sin un alelo CYP2C19 funcional, se recomienda realizar una estrecha monitorización de las reacciones adversas asociadas a voriconazol (ficha técnica de Reyataz®).
42 El efecto resultante de la interacción no está bien establecido (EFV es un inductor moderado del CYP3A4 y un inhibidor de MRP2). Hasta no disponer de más datos, se recomienda evitar la coadministración.
43 Según la ficha técnica europea precaución con inhibidores potentes del CYP3A4. Según la ficha técnica americana evitar la asociación y, en caso de no poder evitarla, reducir la dosis de cabozantinib en 20 mg (por ej: de 60 mg a 40 mg/día o de 40 mg a 20 mg/día) y reanudar la dosis que se usó antes de iniciar el inhibidor del CYP3A4 al cabo de 2 o 3 días después de la interrupción del inhibidor potente del CYP3A4.
44 En cambio no existe riesgo de interacción con lidocaína a dosis bajas para la administración de intramusculares.
46 La levadura de arroz rojo contiene monacolina K (un componente natural con la misma estructura que la lovastatina) que se metaboliza mediante el CYP3A4. Ritonavir, mediante la inhibición potente del CYP3A4, puede aumentar significativamente la concentración de monacolina K y sus efectos adversos como rabdomiólisis. No se recomienda la administración concomitante.
47 Quetiapina: si la coadministración se considera necesaria, valorar reducir la dosis de quetiapina a 1/6 parte.
48 La absorción de tenofovir alafenamida (TAF) puede disminuir durante el tratamiento con RFB debido a la inducción de la P-gp. Sin embargo, estudios con rifampicina y TAF 25 mg/12h o incluso 25 mg/24h sugieren que las concentraciones intracelulares de tenofovir difosfato pueden ser adecuadas (superiores a las alcanzadas con tenofovir disoproxil fumarato en ausencia de rifampicina). No hay estudios con la dosis de 10 mg en presencia de inhibidores enzimáticos, ni datos clínicos con esta combinación.
49 Sonidegib: los inhibidores potentes del CYP3A4 aumentan las concentraciones de sonidegib. La administración concomitante prolongada (más de 14 días) se espera que produzca un aumento mayor de los niveles de sonidegib. En caso de no poder evitar la coadministración, se debe reducir la dosis de sonidegib a 200 mg a días alternos y monitorizar los efectos adversos de sonidegib.
50 Voriconazol: la administración concomitante de dosis estándar de voriconazol con dosis de 400 mg/día de EFV o superiores está contraindicada. La dosis de voriconazol se debe aumentar a 400 mg/12h y la dosis de de EFV se debe reducir a 300 mg/24h (ficha técnica de Vfend®).
51 No deben excederse los 30 mcg/día de etinilestradiol.
52 Valorar RAL 800 mg/12h.
53 Según modelo farmacocinético, concentraciones de nifedipino superiores a las terapéuticas y elevado riesgo de hipotensión.
# En pacientes con neumonía SARS-COV-2 que deban recibir corticoides: el balance riesgo/beneficio del uso de dexametasona durante 10 días en combinación con TAR es favorable. Valorar aumento de dosis de RPV a 50 mg/día y de DOR a 100 mg/12h mientras dure el tratamiento con dexametasona y hasta 2 semanas después de finalizado. En presencia de efavirenz valorar duplicar la dosis de dexametasona. Con TAR potenciado, mantener la dosis habitual y motorizar más estrechamente posible toxicidad por el corticoide.
55 Estos fármacos/plantas son inductores potentes del CYP3A4 y también inducen la UGT, aunque es difícil establecer en qué grado, por lo que es difícil establecer la importancia de la interacción. CAB está contraindicado con inductores potentes de UGT1A1. BIC está contraindicado con inductores potentes a la vez de CYP3A4 y de la UGT1A1. DTG con inductores potentes de CYP3A4 o UGT: en pacientes sin resistencia a INI, aumentar DTG a 50 mg c/12h y en pacientes con resistencia a INI considerar otras alternativas. RAL con inductores potentes de la UGT se puede valorar 800 mg RAL/12h (excepto con RAL 1200mg/24h, que no se recomienda).
56 Si no se puede evitar el uso concomitante de brigatinib con inhibidores potentes de CYP3A, se debe reducir la dosis de brigatinib en un 50 %.
57 Tofacitinib + inhibidores potentes del CYP3A4: reducir la dosis de tofacitinib a 5 mg dos veces al día en pacientes que estén tomando 10 mg dos veces al día y a 5 mg una vez al día en pacientes que estén tomando 5 mg dos veces al día.
58 Solifenacina + inhibidores potentes del CYP3A4: si la asociación se considera necesaria, no superar los 5 mg/día de solifenacina. El tratamiento simultáneo está contraindicado en pacientes con insuficiencia renal grave o insuficiencia hepática moderada.
59 Desfesoterodina/fesoterodina + inhibidores potentes del CYP3A4: si la asociación se considera necesaria, no superar los 4 mg/día de fesoterodina ó 3,5 mg/día de desfesoterodina.
60 Fostamatinib + inhibidores potentes de la CYP3A4: aumenta la exposición del principal metabolito activo, lo que puede elevar el riesgo de reacciones adversas. Monotorizar y ajustar dosis según las indicaciones de ficha técnica.
61 Administrar el ARV inhibidor de glicoproteína‐P separado de afatinib: si el inhibidor de la gp-P se administra c/12h, separar 6h y si el si el inhibidor de la gp-P se administra c/24h, separar 12h.
62 Axitinib + inhibidores potentes del CYP3A4/5: si la asociación se considera necesaria, se recomienda reducir la dosis de axitinib a aproximadamente la mitad de la dosis (Ej., la dosis de inicio debería reducirse de 5 mg dos veces al día a 2 mg dos veces al día). Si se interrumpe la administración conjunta del inhibidor potente, debería considerarse la vuelta a la dosis de axitinib utilizada de forma previa al inicio del inhibidor potente del CYP3A4/5.
63 Ruxolitinib: cuando se administra con inhibidores potentes del CYP3A4 o inhibidores duales de las enzimas CYP2C9 y CYP3A4 como fluconazol, idosis de ruxolitinib aprox. 50% y administrarlo dos veces al día. Evitar el uso concomitante de ruxolitinib con dosis diarias de fluconazol mayores a 200 mg.
64 Lorlatinib + inhibidores potentes del CYP3A4/5: si la asociación se considera necesaria, la dosis inicial de lorlatinib de 100 mg c/24h se debe reducir a 75 mg c/24h. Si se suspende el uso concomitante del inhibidor potente del CYP3A4/5, se debe reanudar el tratamiento con lorlatinib a la dosis utilizada anteriormente.
65 Monitorizar concentraciones plasmáticas y ajustar la dosis.
66 El fármaco es un inductor potente del CYP3A4. No hay datos sobre su posible efecto sobre la glucuronidación. Evitar la asociación en la medida de lo posible.
Bibliografía:
Plan Nacional sobre el Sida (SPNS), Grupo de Estudio de Sida (GeSIDA) y Sociedad Española de Farmacia Hospitalaria (SEFH). Documento de consenso para mejorar la adherencia a la farmacoterapia en pacientes con infección por el virus de la inmunodeficiencia humana en tratamiento antirretroviral (Actualización febrero 2020). https://mscbs.gob.es/ciudadanos/enfLesiones/enfTransmisibles/sida/publicaciones/ profSanitarios/docAdherencia_actualizacionFeb20.pdf (consultada 28.02.2020).
Quintana Y, Gonzalez Martorell EA, Fahy D, et al. A systematic review on promoting adherence to antiretroviral therapy in HIV-infected patients using mobile phone Appl Clin Inform 2018; 09:450-66.
Kanters S, Park JJ, Chan K, et al. Interventions to improve adherence to antiretroviral therapy: a systematic review and network meta-analysis. The Lancet HIV 2017; 4:e31-e40.
Calin R, Landowski S, Valantin MA, et al. Efficacy of intermittent short cycles of integrase inhibitor-based maintenance ART in virologically suppressed HIV patients. J Antimicrob Chemother 2020; 75:1321-3.
Castillo-Mancilla JR, Brown TT, Erlandson KM, et Suboptimal adherence to combination antiretroviral therapy is associated with higher levels of inflammation despite HIV suppression. Clin Infect Dis 2016; 63:1661-7.
Sutton SS, Magagnoli J, Hardin JW. Odds of viral suppression by single-tablet regimens, multiple-tablet regimens, and adherence level in HIV/AIDS patients receiving antiretroviral Pharmacotherapy 2017; 37:204-13.
Scarsi KK, Swindells S. The Promise of improved adherence with long-acting antiretroviral therapy: What are the data? J Int Assoc Prov AIDS Care 2021; 20:1-10.
Murray MM, Lin J, Buros Stein A, et al. Relationship of polypharmacy to HIV RNA suppression in people aged 50 years living with HIV. HIV Med 2021 Jun 2. doi: 10.1111/ 13122. Online ahead of print.
Desprescripción farmacológica de la terapia no antirretroviral en pacientes con infección por VIH (Actualización Octubre de 2018). Panel de expertos de GeSIDA. http://gesida- org/wp-content/uploads/2019/03/15_DESPRESCRIPCION_FARMACOLOGICA_ TERAPIA_NO_ANTIRRETROVIRAL_EN_PACIENTES_CON_INFECCION_POR_VIH.pdf (Consultada 30.11.2021).
Llibre JM, Arribas JR, Domingo P, et al. Clinical implications of fixed-dose coformulations of antiretrovirals on the outcome of HIV-1 AIDS 2011; 25:1683-90.
Sebaaly JC, Kelley D. Single-tablet regimens for the treatment of HIV-1 infection. Ann Pharmacother 2017; 51:332-44.
Li H, Marley G, Ma W, et al. The role of ARV associated adverse drug reactions in influencing adherence among HIV-infected individuals: a systematic review and qualitative meta-synthesis. AIDS Behav 2017; 21:341-51.
National HIV Antiretroviral therapy. Adverse effects of antiretroviral medications. Disponible en: www.hiv.uw.edu/go/antiretroviral-therapy/adverse-effects/core-concept/all
Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 screening for hypersensitivity to N Engl J Med 2008; 358:568-79.
Hoffmann C, Welz T, Sabranski M, et al. Higher rates of neuropsychiatric adverse events leading to dolutegravir discontinuation in women and older patients. HIV Med 2017; 18:56-63.
Peñafiel J, de Lazzari E, Padilla M, et Tolerability of integrase inhibitors in a real-life setting. J Antimicrob Chemother 2017; 72:1752-9.
Wohl D, Clarke A, Maggiolo F, et al. Patient-reported symptoms over 48 weeks among participants in randomized, double-blind, phase III non-inferiority trials of adults with HIV on co-formulated bictegravir, emtricitabine, and tenofovir alafenamide versus co-formulated abacavir, dolutegravir, and lamivudine. Patient 2018; 11:561-73.
Jaeger H, Overton ET, Richmond G et al. Week 96 efficacy and safety of long acting cabotegravir + rilpivirine every two months: ATLAS 2M. Conference on Retroviruses and Opportunistic Infections, March, 6-10, 2021; Virtual.
Orkin C, Bernal Morell E, Tan DHS, et al. Initiation of long-acting cabotegravir plus rilpivirine as direct-to-injection or with an oral lead-in in adults with HIV-1 infection: week 124 results of the open-label phase 3 FLAIR Lancet HIV 2021;8:e668–e678.
Buzón-Martín L. Weight gain in HIV-infected individuals using distinct antiretroviral AIDS Rev 2020; 22:158-67.
Venter WDF, Moorhouse M, Sokhela S, Fairlie L, Mashabane N, Masenya M, et al. Dolutegravir plus two different prodrugs of tenofovir to treat HIV. N Engl J Med 2019; 381:803-15.
Swindells S, Andrade-Villanueva JF, Richmond GJ, et al. Long-acting cabotegravir and rilpivirine for maintenance of HIV-1 suppression. N Engl J Med 2020; 382:1112-23.
Orkin C, Arasteh K, Hernandez-Mora MG, et al. Long-acting cabotegravir and rilpivirine after oral induction for HIV-1 N Engl J Med 2020; 382:1124-5.
Lee FJ, Monteiro P, Baker D, et al. Rosuvastatin vs. protease inhibitor switching for hypercholesterolaemia: a randomized HIV Med 2016; 17:605-14.
Hoy J, Richardson R, Ebeling PR, et al. Zoledronic acid is superior to tenofovir disoproxil fumarate-switching for low bone mineral density in adults with HIV. AIDS 2018; 32:1967-75.
Deutschmann E, Bucher HC, Jaeckel S, et al. Prevalence of potential drug-drug interactions in patients of the Swiss HIV Cohort Study in the era of HIV integrase inhibitors [published online ahead of print, 2020 Jul 8]. Clin Infect Dis 2020; ciaa918. doi:10.1093/ cid/ciaa918.
López-Centeno B, Badenes-Olmedo C, Mataix-Sanjuan Á, et al. Polypharmacy and drug-drug interactions in HIV-infected subjects in the region of Madrid, Spain: a population-based study. Clin Infect Dis 2020; 71:353-62. doi:10.1093/cid/ciz811.
Molas E, Luque S, Retamero A, et Frequency and severity of potential drug interactions in a cohort of HIV-infected patients identified through a multidisciplinary team. HIV Clin Trials 2018;19:1-7. doi:10.1080/15284336.2017.1404690.
Demessine L, Payro-Saint paul L, Gardner EM, et al. Risk and cost associated with drug-drug interactions among aging HIV patients receiving combined antiretroviral therapy in France. Open Forum Infect Dis 2019;6:ofz051. doi: 10.1093/ofid/ofz051.
Bracchi M, Stuart D, Castles R, Khoo S, Back D, Boffito M. Increasing use of 'party drugs' in people living with HIV on antiretrovirals: a concern for patient AIDS 2015;29:1585-92. doi:10.1097/QAD.0000000000000786.
Lopes S, O'Day K, Meyer K, et Comedication prescription patterns and potential for drug-drug interactions with antiretroviral therapy in people living with human immunodeficiency virus type 1 infection in Germany. Pharmacoepidemiol Drug Saf 2020; 29:270-8. doi:10.1002/pds.4928.
Umland T, Stellbrink HJ, Calvo M, Zahn A et The clinical relevance of potential drug- drug interactions (DDIs) with bictegravir/emtricitabine/tenofovir alafenamide (B/F/TAF) – Real-world data from the German IQVIA prescription database. P100. Presentado en: HIV Glasgow, 05-08 October 2020, Glasgow, UK. Disponible en: http://www.hivglasgow.org/ wp-content/uploads/2020/11/P100_HIVGlasgowBictarvyIQVIA.pdf. Acceso: septiembre 2021.
Nhean S, Tseng A, Back The intersection of drug interactions and adverse reactions in contemporary antiretroviral therapy [published online ahead of print, 2021 Aug 26]. Curr Opin HIV AIDS 2021;doi: 10.1097/COH.0000000000000701.
Hodge D, Back DJ, Gibbons S, Khoo SH, Marzolini C. Pharmacokinetics and drug-drug interactions of long-acting intramuscular cabotegravir and rilpivirine. Clin Pharmacokinet 2021; 60:835-53. doi:10.1007/s40262-021-01005-1.
Cattaneo et Lack of clinically relevant interactions between bictegravir and metformin in persons with diabetes and HIV. J Antimicrob Chemother 2021; 76:1945-6.doi: 10.1093/ jac/dkab077.
Gervasoni C, Formenti T, Cattaneo D. Management of polypharmacy and drug-drug interactions in HIV patients: a 2-year experience of a multidisciplinary outpatient clinic. AIDS Rev 2019; 21:40-9. doi:10.24875/AIDSRev.19000035.
7. SITUACIONES ESPECIALES
7.1. Primoinfección por el VIH-1.
7.2. Infección por VIH-2
7.3. Embarazo
7.4. Comorbilidades
Primoinfección por el VIH-1.
La primoinfección por el VIH-1 se asocia con síntomas inespecíficos (fiebre, cefalea y/o malestar general) y banales en la mayoría de casos, por lo que con frecuencia puede pasar desapercibida, aunque algunos pacientes presentan un cuadro similar al de la mononucleosis infecciosa12. Las pruebas de ELISA de cuarta generación, que incluyen la determinación simultánea de anticuerpos y del antígeno p24 del VIH-1, permiten el diagnóstico durante la infección aguda en el 80-90% de casos. Se considera infección aguda a aquella de menos de 30 días e infección reciente a la de menos de 180 días12.
Se recomienda el inicio de TAR en todos los casos de primoinfección12 ya que acorta la duración y gravedad de los síntomas, suprime rápidamente la replicación viral, reduce la diversidad viral y el reservorio (ADN proviral), normaliza más rápidamente la cifra de linfocitos CD4+ y el cociente CD4/CD8, reduce la activación inmunológica, preserva o restaura la inmunidad específica frente al VIH-1 y reduce la transmisión3.
Una vez iniciado el TAR durante la primoinfección, no se recomienda su interrupción, ya que ninguna estrategia evaluada (tratamiento intermitente, vacunas terapéuticas o imunomoduladores adyuvantes) ha permitido evitar el rebote viral en la gran mayoría de pacientes4. En muy pocos pacientes que iniciaron el TAR precozmente (<90 días) se observó un control virológico cuando lo interrumpieron (controladores post-tratamiento), pero no se han establecido parámetros para identificarlos basalmente5. Por tanto, en la práctica clínica, se debe iniciar el TAR durante la primoinfección y se debe mantener de forma indefinida, como en la infección crónica. Las pautas de TAR deben ser las mismas que en la infección crónica y la adición de más FAR (4 ó 5) no está recomendada, en general, ya que no se ha acompañado de ningún beneficio adicional6. Casi no hay experiencia con biterapia.
En los pacientes gravemente sintomáticos, el inicio del TAR debe realizarse de forma inmediata (idealmente en la primera visita, si se considera que el paciente está preparado para ello), ya que acorta la duración y gravedad de los síntomas. En el resto de pacientes debe iniciarse lo antes posible, ya que es cuando se puede conseguir el máximo beneficio inmunológico (>900 linfocitos CD4+/μL y ratio CD4/CD8 normal)7. En cada región se deben conocer las tasas de transmisión de virus resistentes.
En usuarios de PrEP con TDF/FTC que se infectan por el VIH-1, la tasa de resistencias es baja. Sin embargo, cuando la PrEP se ha iniciado de forma inadvertida en la fase inicial de una infección aguda (Fiebig I, solo RNA detectable, y tanto anticuerpos como Ag P24 negativos), pueden atenuarse marcadamente las manifestaciones clínicas y en hasta cerca de la mitad de los casos desarrollarse MR al FTC y en menor medida al TDF, lo que debería tenerse en cuenta en la adaptación del TAR inicial y considerar la adición de un cuarto fármaco, a la espera del test de resistencia. Excepcionalmente, puede producirse el contagio por exposición a una fuente con una cepa resistente a los componentes de la PrEP8.
Finalmente, siempre debe valorarse la inclusión de estos pacientes, en especial de aquellos que llevan muy pocos días infectados, en protocolos de investigación o ensayos clínicos que busquen la erradicación o la cura funcional del VIH-1.
Recomendaciones
- El TAR debe recomendarse en todos los pacientes con primoinfección por el VIH-1 independientemente de los síntomas, gravedad y duración; para obtener el máximo beneficio debe comenzarse tan pronto como sea posible (A-II).
- El tipo de TAR será el mismo que en la infección crónica (A-I), basado en un INI de alta barrera genética (DTG o BIC) ya que reducen la CVP más rápido que otras familias, pero no una pauta doble.
- TDF o TAF/FTC serán los ITIAN recomendados si no se cuenta con serología de VHB y HLA-B5701 (A-I).
- Si es una primoinfección en un usuario reciente de PrEP se podría añadir un cuarto fármaco (DRV/c) hasta disponer del resultado del test de resistencias (C-III).
- Una vez iniciado el TAR debe mantenerse indefinidamente (A-I).
Infección por VIH-2
La infección por el VIH-2 es endémica en África Occidental, con prevalencias superiores al 1% en algunas zonas de Guinea-Bissau y Costa de Marfil. La globalización ha dado lugar a un número considerable de casos en otras partes de África, Europa, India y Brasil. Se estima que entre 1 y 2 millones de personas están infectadas por VIH-2. En España la prevalencia es muy baja, aunque debe descartarse en pacientes procedentes de zonas endémicas o que hayan sido parejas de personas de esas regiones1.
A diferencia del VIH-1: 1) La infección por VIH-2 se caracteriza por un estadio asintomático más largo, con valores de CVP más bajos y con frecuencia indetectables. 2) No disponemos de pruebas comerciales para medir la CVP, ni el estudio de resistencias genotípicas en VIH-2, aunque algunos laboratorios han desarrollado procedimientos no comerciales que han sido suficientemente validados12. 3) Los algoritmos genotípicos utilizados para predecir resistencias a TAR en el VIH-1 no son directamente aplicables al VIH-22. 4) Cuando la inmunodeficiencia avanza, los pacientes con infección por el VIH-2 se recuperan con más dificultad, con peor respuesta al TAR y normalización de linfocitos CD4+.
Los principios generales del TAR en pacientes con infección por VIH-2 deben ser los mismos que para la infección por VIH-1. El VIH-2 presenta resistencia intrínseca a los ITINN y una sensibilidad variable frente a los IP, siendo DRV el más activo. Los INI son
activos frente al VIH-234. Ibalizumab ha demostrado actividad in vitro frente a aislados de VIH-2 del grupo A y del B5.
Recomendaciones
- Se aconseja monitorizar clínica y CD4+ cada 6-12 meses y, si está disponible, la CVP de VIH-2 (A-III).
- El uso de ITINN está contraindicado en el tratamiento de la infección por VIH-2
- (A-I).
- El régimen de TAR de inicio en estos pacientes es la combinación de 2 ITIAN + 1 INI (A-II). Como alternativa a los INI, puede aconsejarse DRV/c.
- En pacientes con infección dual VIH-1/VIH-2 el TAR debe seguir las recomendaciones del VIH-2 (A-III).
Embarazo
El TAR en el embarazo se discute en un documento de consenso elaborado por el PNS en colaboración con GeSIDA y otras sociedades pendiente de actualización. Se recomienda la lectura de guías actualizadas1 ante cualquier duda al respecto.
Este apartado presenta las recomendaciones acerca del TAR en este contexto. En las (Tablas 10 y 11) se recoge la actitud recomendada ante diferentes situaciones.
El objetivo del TAR durante el embarazo es conseguir y mantener CVP indetectable durante el mayor tiempo posible, especialmente en el tercer trimestre y en el momento del parto2.
El TAR de elección en la mujer con deseo de gestación o gestante se basa en la combinación de un INI, de elección RAL o DTG, o como alternativa DRV/r, junto a 2
ITIAN, de elección ABC/3TC o TDF/FTC o TAF/FTC)(1,3-5).
Recomendaciones
- Si el diagnóstico de infección por el VIH-1 se realiza durante el embarazo, el TAR se comenzará lo más precozmente posible, y preferiblemente con un régimen que incluya INI, dada la posibilidad de transmisión intrauterina (A-I).
- La elección de los FAR concretos se basará en el estudio de resistencias y en la seguridad de los mismos (Tablas 10 y 11). El TAR de elección es TDF/FTC, TAF/ FTC o ABC/3TC, con RAL 400 mg dos veces al día (A-I), DTG una vez al día (A-II) o DRV/r dos veces al día (A-II). Podrán recibir cualquiera de los FAR “recomendados” o “alternativos” tras una valoración individualizada (A-III).
- En caso de que una mujer esté recibiendo biterapia y se quede embarazada, se recomienda el cambio a triple terapia (C-III).
- El tratamiento intraparto con ZDV vía intravenosa estará indicado, independientemente del TAR que llevase o hubiese llevado, si la CVP en el parto es >1000 cop/mL o desconocida (A-I); entre 50 y 999 cop/mL también se recomienda (B-III).
Comorbilidades
7.4.1. TRATAMIENTO ANTIRRETROVIRAL DE INICIO EN PACIENTES CON INFECCIONES OPORTUNISTAS (IO) DISTINTAS A LA TUBERCULOSIS (TB).
El momento idóneo para iniciar el TAR en un paciente con una IO es motivo de controversia1. Las posibles ventajas de un inicio temprano incluyen una recuperación inmune más rápida, una mayor resolución de la IO, prevenir la aparición de otras y reducir el riesgo de mortalidad. Entre los inconvenientes destacan las posibles interacciones y toxicidades y el síndrome inflamatorio de reconstitución inmune (SIRI)1. El ensayo clínico aleatorizado ACTG A5164 demostró una reducción en la progresión a SIDA/muerte (HR: 0,53; IC 95%: 0,30 a 0,92) en pacientes que iniciaban TAR dentro de una mediana de 12 días tras el inicio del tratamiento de la IO frente a demorarlo hasta una mediana de 45 días2. No se observaron diferencias en efectos adversos ni en la incidencia de SIRI, según el uso de esteroides para la IO. Más del 60% de los pacientes presentaban neumonía por Pneumocystis jirovecii (NPJ), pero dado el pequeño número y variedad de otras IO es difícil extraer conclusiones para cada una de ellas por separado.
El ensayo clínico IDEAL, ha comparado una estrategia de inicio inmediato del TAR (7 primeros días tras iniciar tratamiento para la IO) frente a un inicio diferido (21-42 días)3. Se incluyeron pacientes con encefalitis por toxoplasma (11 sujetos) y NPJ (50 sujetos). Todos los sujetos iniciaron tratamiento con TDF+FTC+ATV/r. El ensayo no tuvo potencia suficiente para detectar diferencias en ninguno de los “end point” primarios o secundarios entre los dos grupos. Los autores concluyen que el inicio inmediato de TAR parece seguro y no muestra más probabilidad de progresión de IO, SIRI, fracaso inmunovirológico o peor calidad de vida.
Una de las IO más complicadas de tratar por su elevada morbimortalidad es la meningitis criptocócica. En el ensayo clínico COAT4, se incluyeron 177 sujetos y fueron aleatorizados a inicio precoz o tardío del TAR (1-2 semanas o 5 semanas después del diagnóstico). La mortalidad a las 26 semanas fue mayor en los que iniciaron el TAR precozmente (45% vs. 30%; RR:1,73; IC95%:1,06 a 2,82, p=0,03). La presencia de SIRI no difirió entre ambos grupos (20% vs. 13%, p=0,32)4. Estos resultados se confirmaron en una revisión sistemática de la colaboración Cochrane que incluía 4 estudios (n=294 pacientes)5. El riesgo de muerte se incrementaba en un 42% cuando se iniciaba el TAR de forma precoz vs tardío (RR 1,42; IC95% 1,02- 1,97). Este incremento en la mortalidad no parecía estar claramente relacionado con el SIRI.
Por tanto, a pesar de la limitación de los datos, en las IO en general habría una menor progresión de la enfermedad en aquellos pacientes que inician el TAR de forma precoz (en los primeros 14 días), aunque no está claro si, además de la NPJ, esto es cierto para todas las IO. Concretamente, en la meningitis criptocócica sería mejor esperar varias semanas para iniciar el TAR tras el diagnóstico y tratamiento de esta IO45. En caso de presentar dos o más IO simultáneamente, siendo una de ellas una meningitis criptocócica o tuberculosa, el buen juicio clínico debe sopesar el beneficio o perjuicio de iniciar TAR precoz (< 2 semanas), intermedio (2-5 semanas) o tardío (> 5 semanas).
Recomendaciones
- Sin considerar la TB, en la mayoría de las IO, excepto la meningitis criptocócica, se debe iniciar el TAR lo antes posible (se recomienda en las dos primeras semanas tras el inicio del tratamiento de la IO) (A-II).
- En pacientes con NPJ que no reciben TAR, éste se debería comenzar en las dos primeras semanas tras el diagnóstico de la NPJ (A-I).
- En pacientes con meningitis criptocócica se recomienda diferir el inicio del TAR 5 semanas por el riesgo de mayor mortalidad asociada con un inicio precoz del mismo (especialmente en pacientes con menos de 5 células/μL en el LCR o incremento de la presión intracraneal) (A-I).
7.4.2. TRATAMIENTO ANTIRRETROVIRAL Y TUBERCULOSIS (TB).
El tratamiento de la TB en adultos con infección por el VIH-1 ha sido objeto de un documento de consenso específico de GeSIDA/Secretaría del PNS1. Se recomienda su lectura.
Momento óptimo de inicio del TAR en pacientes infectados por el VIH-1 con TB1
Recomendaciones
- Se recomienda iniciar TAR independientemente de la cifra de CD4+ en las dos primeras semanas, una vez comprobada la tolerancia al tratamiento antituberculoso (A-I).
- En el caso de la meningitis tuberculosa, se recomienda demorar el inicio del TAR al menos 4 semanas, eligiendo el momento óptimo en función de la situación clínica del paciente (A-I).
- Con las recomendaciones previas se disminuye el riesgo de efectos adversos y el desarrollo de SIRI, sin comprometer la supervivencia.
Pautas de TAR.
La principal dificultad radica en las posibles interacciones medicamentosas con los fármacos antituberculosos, especialmente relevantes en el caso de las rifamicinas.
Tanto para los pacientes que inician TAR como para los pacientes que ya están recibiendo TAR, el régimen debe construirse de acuerdo a los principios para la elección de FAR en pacientes en tratamiento para TB que se sintetizan a continuación. Además, hay que tener en cuenta los antecedentes de resistencias o intolerancias a FAR.
- Elección de los ITIAN. No existe interacción significativa entre los fármacos antituberculosos y la mayoría de ITIAN, ni hay evidencia de la potenciación de la toxicidad entre ellos. Una excepción la constituye TAF porque la rifampicina disminuye su concentración plasmática un 55%.
- Elección del tercer fármaco. La mayor experiencia y los mejores resultados se han obtenido con EFV, por lo que en el caso de los pacientes con TB, sigue constituyendo el fármaco de elección. También puede utilizarse en el caso de necesitar dosis muy altas de rifampicina para tratar meningitis2.
- Alternativas terapéuticas como terceros fármacos. Existen datos para recomendar como alternativas pautas que incluyan RAL 800 mg/12 horas3 o DTG 50 mg/12 horas4. RAL 400 mg/12 horas no ha demostrado la no inferioridad frente a EFV5. Con las alternativas actualmente disponibles no se recomienda la administración de MVC salvo en casos Si no existen alternativas, CAB oral podría coadminstrarse con rifabutina, aunque todavía falta evidencia para recomendar su uso6.
- Fármacos que no pueden No se pueden administrar con rifampicina los ITINN diferentes de EFV (RPV, ETR y DOR), ningún IP (potenciado o no), ni EVG, RAL 1200 mg QD, BIC, CAB6, ni fostemsavir.
Recomendaciones
- Elección de los ITIAN. Se puede utilizar ABC, TDF, 3TC o FTC (A-I). No está recomendado el uso de TAF con rifampicina (A-II).
- Elección del tercer fármaco. Se recomienda el uso de EFV a dosis estándar (A-I). Como alternativas se recomiendan RAL 800 mg/12 h o DTG 50 mg/12 h (A-II) y en el caso excepcional de que la única opción fuese un IP debe sustituirse rifampicina por rifabutina y realizar el ajuste de dosis correspondiente (A-I).
Síndrome inflamatorio por reconstitución inmunológica (SIRI).
El SIRI es una complicación frecuente del TAR en pacientes con TB, especialmente en pacientes con recuento de células CD4+ muy bajos, cuando el TAR se inicia muy precozmente en relación con el inicio del tratamiento antituberculoso. En pacientes inmunodeprimidos, el uso de prednisona puede prevenir el desarrollo de SIRI7. Un ensayo aleatorizado, doble ciego, controlado con placebo de prednisona (1,5 mg/kg/día durante 2 semanas y luego 0,75 mg/kg/día durante 2 semanas), en el que se incluyeron 55 pacientes por rama, la prednisona redujo la necesidad de hospitalización y procedimientos terapéuticos y aceleró la mejoría clínica de los pacientes.
Recomendaciones
- En caso de SIRI no debe interrumpirse el tratamiento antituberculoso ni el TAR (A-III).
- En pacientes con TB y un recuento de linfocitos CD4+ menor de 100 células/ μL se recomienda administrar prednisona (40 mg/día durante 2 semanas y luego 20 mg/día durante 2 semanas más) para prevenir el desarrollo de SIRI (A-I).
- Una vez desarrollado SIRI, para el manejo de los síntomas pueden añadirse antiinflamatorios no esteroideos en las formas leves o moderadas (A-III) o corticosteroides en las formas graves (A-II).
7.4.3. USO DE TAR EN PACIENTES CON INSUFICIENCIA RENAL
La insuficiencia renal crónica en la población infectada por el VIH-1 tiene una prevalencia variable en relación con la población estudiada(!). El método considerado de elección en la actualidad para estimar la función renal es la ecuación CKD- EPI2. Para una visión completa del diagnóstico, prevención y tratamiento de las alteraciones renales en pacientes con infección por el VIH se recomienda consultar el documento de consenso ad hoc elaborado por GeSIDA3. La dosificación de los fármacos según el FGe se expone en la Tabla 2.
Recomendaciones
- Es necesario ajustar las dosis de los ITIAN, excepto en el caso de ABC (A-II). MVC requiere ajuste de dosis si se emplea en combinación con inhibidores potentes del CYP3A4, como los IP, ketoconazol, itraconazol, claritromicina y telitromicina (A-II).
- No se requiere ajuste de dosis para los ITINN (en pacientes con insuficiencia renal avanzada en tratamiento con RPV se recomienda vigilancia estrecha por el potencial aumento de sus concentraciones plasmáticas), IP, los inhibidores de la entrada, ibalizumab y fostemsavir, ni para los INI RAL, DTG o CAB (A-II).
- En general, se desaconseja el uso de coformulaciones de FAR en los que alguno de los fármacos precise ajuste de dosis en pacientes con insuficiencia renal significativa. TDF/FTC/c/EVG no debe emplearse en pacientes con FGe
- <70 ml/min. TDF/FTC/EFV y TDF/FTC/RPV no deben emplearse en pacientes con FGe <50 ml/min y DTG/3TC/ABC, DTG/3TC, TAF/FTC/c/EVG, TAF/FTC/ BIC, TAF/FTC/RPV y TAF/FTC/c/DRV no deben utilizarse en pacientes con FGe <30 ml/min. En estos casos, deben emplearse los FAR por separado y realizar los ajustes pertinentes (B-II). DTG/RPV y CAB+RPV deben usarse con precaución en pacientes con FGe <30 ml/min (B-III).
- En los pacientes con cualquier grado de insuficiencia renal se recomienda vigilar estrechamente la función renal, incluyendo la función tubular en el caso de utilizar TDF y evitar los fármacos nefrotóxicos (A-III).
- En los pacientes con insuficiencia renal crónica avanzada se debe realizar el ajuste de dosis recomendado por las fichas técnicas de cada medicamento, teniendo en cuenta las posibles interacciones entre los diferentes fármacos, más frecuentes y peligrosas en esta situación (A-II). Si no existen contraindicaciones, se puede utilizar la combinación de ABC más 3TC (ajustado al FGe) con un ITINN, un INI no potenciado (DTG o RAL), DRV/r o DRV/c. También biterapia con 3TC ajustado al FGe con DTG, DRV/r o DRV/c (A-III).
7.4.4. HEPATOPATÍAS (VHC, VHB, CIRROSIS)
La AEEH y la SEIMC han publicado una guía de manejo de la hepatitis C, que incluye al paciente coinfectado1, la cual recomendamos consultar para más detalles. Entre las causas principales de daño hepático en los pacientes con infección por el VIH-1 actualmente están la hepatitis B crónica y la enfermedad hepática grasa2345. La hepatitis B crónica mantiene una prevalencia estable (3,2%) y causa cirrosis en a la esteatosis/esteatohepatitis no alcohólica, es una causa cada vez más frecuente de hepatopatía y está cobrando mayor relevancia con la menor prevalencia de la hepatitis vírica y con el envejecimiento de la población infectada por VIH34.
Recomendaciones
- El cuidado y seguimiento del paciente con daño hepático avanzado se debe de hacer como en la población general y se aconseja seguir las recomendaciones de las guías clínicas existentes (A-III).
- Se debe realizar despistaje de la enfermedad hepática grasa en todo paciente infectado por VIH-1 con sospecha de hepatopatía, obesidad o síndrome metabólico (A-II).
7.4.4.1. Cuándo iniciar el TAR en pacientes coinfectados
En estudios de cohortes, el control de la replicación del VIH-1 y la mejoría inmunológica por efecto del TAR se han asociado a una menor velocidad de progresión de la enfermedad hepática causada por VHC y a una mejor evolución clínica, incluso en pacientes con cirrosis descompensada5. Por tanto, es preferible iniciar el TAR y, una vez controlado el VIH-1, iniciar el tratamiento de la hepatitis. Opcionalmente, en pacientes controladores de élite, se podría plantear tratar inicialmente el VHC.
Tanto TDF como TAF suprimen el VHB en la mayoría de los pacientes coinfectados por VIH-1 y VHB6789. La retirada de un tratamiento eficaz frente al VHB puede dar lugar a una exacerbación de la hepatitis con consecuencias potencialmente graves y es de especial importancia tenerlo presente en las simplificaciones a biterapias10.
Recomendaciones
- En pacientes coinfectados por VHB o VHC se recomienda iniciar el TAR tan pronto como sea posible (A-I).
- En pacientes que precisan tratamiento de la hepatitis C, en general es preferible iniciar primero el TAR (B-III).
- En pacientes coinfectados por VIH/VHB se debe iniciar precozmente un TAR que incluya TDF o TAF y FTC o 3TC (A-I).
7.4.4.2. Elección de los fármacos antirretrovirales (FAR)
La elección de los FAR en un paciente coinfectado por virus de la hepatitis ha de tener en cuenta su potencial hepatotoxicidad, la existencia de cirrosis hepática y si la coinfección es por VHB y/o VHC y/o VHD.
En cuanto a la hepatotoxicidad, los FAR utilizados actualmente tienen bajo riesgo y la mayoría de los episodios son leves y autolimitados.
Por otra parte, no se ha demostrado que los FAR ejerzan un efecto protector específico sobre la fibrogénesis hepática. En pacientes con esteatosis hepática en tratamiento con EFV, un ensayo piloto randomizado mostró que los que eran aleatorizados a RAL experimentaban un descenso de los niveles de esteatosis11.
En pacientes con cirrosis estadio A de Child-Pugh se puede usar cualquier FAR, con las consideraciones antes expuestas. En pacientes con insuficiencia hepatocelular (estadios B y C de Child-Pugh) existen, en general, pocos datos Tabla 1.
Por sus características farmacocinéticas, los INI ofrecen ventajas en estos pacientes. En estadios A y B de Child-Pugh no requieren ajuste de dosis1213. En estadio C, RAL ha demostrado buena tolerabilidad14 y su metabolismo no depende del citocromo P450; el metabolismo de DTG, BIC y CAB depende sólo en parte del citocromo CYP3A4 lo que les hace fármacos de bajo riesgo.
Existen múltiples interacciones entre los FAR y los antivirales de acción directa (AAD) frente al VHC que pueden condicionar una modificación de la dosis o desaconsejar su coadministración y debe tenerse siempre en cuenta (https:// www.hiv-druginteractions.org)15.
Recomendaciones
- En pacientes con hepatopatía crónica y función hepática conservada, incluida la cirrosis estadio A de Child-Pugh se puede utilizar cualquier FAR (A-I).
- En pacientes con insuficiencia hepatocelular leve/moderada (Child- Pugh A o B), los ITIAN (salvo ABC), los INI, RPV, DOR o DRV no precisan ajuste de dosis y son fármacos de elección (A-II).
- En pacientes en estadio de Child-Pugh C, se deben considerar como pautas preferentes aquellas basadas en RAL (A-II), DTG y BIC (B-III).
- Antes de prescribir un tratamiento con AAD en un paciente que recibe TAR, debe consultarse una aplicación informática actualizada de interacciones farmacológicas (A-III).
- En pacientes coinfectados por VIH/VHB se debe evitar la interrupción de una pauta eficaz frente a VHB (A-II).
7.4.5. NEOPLASIAS
Se recomienda la lectura de documentos específicos acerca de las neoplasias en pacientes con infección por el VIH-11234. En general, el TAR debe iniciarse lo antes posible. Se puede considerar una diferencia de al menos 7 días respecto al inicio del tratamiento antineoplásico para asegurar la tolerancia. En los pacientes en TAR pueden precisarse cambios dependiendo de las interacciones y la toxicidad añadida.
Para seleccionar los FAR es importante considerar el perfil de toxicidad y las posibles interacciones farmacocinéticas con los fármacos antineoplásicos, principalmente de aquellos FAR que se metabolizan por la vía del CYP45021. Los IP/p son los FAR que presentan interacciones farmacológicas más importantes. Se aconseja consultar páginas web sobre interacciones que se actualicen con regularidad.
Los INI no potenciados respecto a los IP/p e ITINN tienen, en general, ventajas en sus características farmacológicas, perfil de tolerancia e interacciones para el tratamiento concomitante con fármacos antineoplásicos1.
Los IP/p se asocian a neutropenia más profunda y prolongada cuando se coadministran con la quimioterapia de los linfomas12. Algunos de ellos, así como la RPV, alargan el intervalo QT, lo que podría potenciar este efecto cuando se usan junto a determinados fármacos antineoplásicos1. Además, los IP/p pueden presentar mayor toxicidad, más interacciones y menor eficacia virológica que los ITINN y RAL5.
Recomendaciones
El TAR es un pilar fundamental, y debe formar parte, del tratamiento de los pacientes con neoplasias e infección por el VIH-1 (A-II).
RAL y DTG, por sus características farmacológicas, excelente tolerancia y mínimas interacciones, deben ser los FAR de elección en pacientes que reciban quimioterapia (A-II). Como alternativa, se puede considerar el uso de BIC y DOR (C-III).
Tablas:
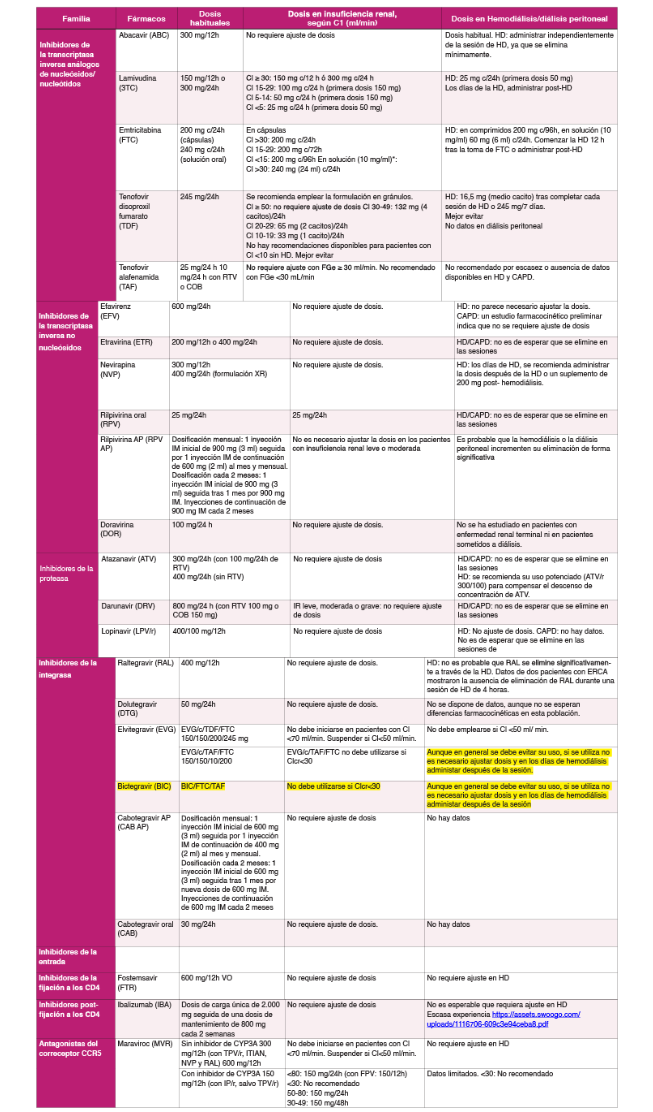
*Las cápsulas y la solución oral de emtricitabina tienen diferente biodisponibilidad, de forma que con 240 mg de la solución oral (24 ml) se alcanzan unas
concentraciones plasmáticas similares a las alcanzadas con 200 mg en cápsulas.
CH: cirrosis hepática; Cl: aclaramiento de creatinina en ml/min; ERCA: enfermedad renal crónica avanzada, HD: hemodiálisis; CAPD: diálisis peritoneal; IH: insuficiencia hepática; IR: insuficiencia renal; MATE1: transportador de expulsión de toxinas y multifármacos 1; OAT1 y 3: transportadores de aniones orgánicos 1 y 3; OCT2: transportador de cationes orgánicos 2;AP, acción prolongada.
*Con el término de TDF nos referimos a cualquiera de las sales de tenofovir disponibles (fumarato, fosfato, maleato, succinato).
1Si en esta situación, la presentación es próxima al parto, se puede valorar también la adición de RAL a dosis de 400 mg/12 horas (siempre y cuando no se sospeche resistencia y no se hubiera utilizado previamente) dado su rápido paso placentario.
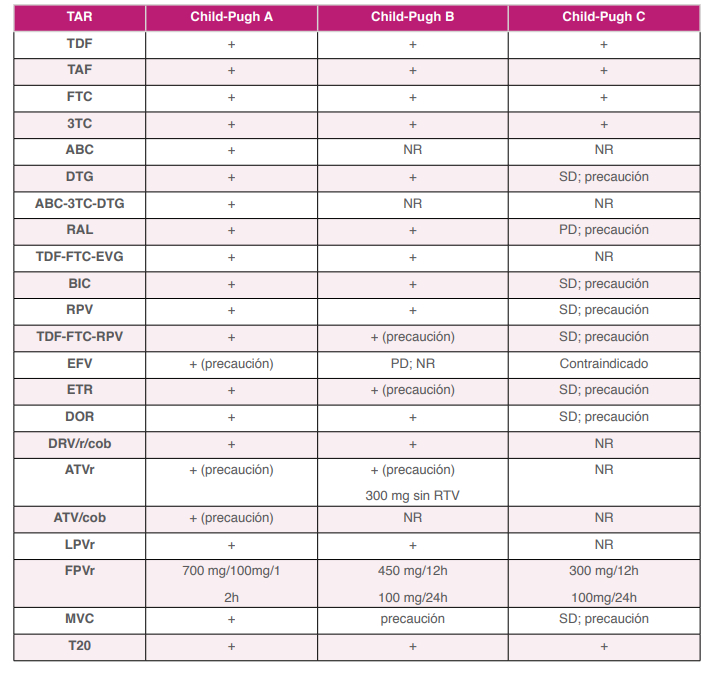
+: Fármaco seguro, no precisa ajuste de dosis.
PD: Pocos datos.
SD: sin datos suficientes sobre la dosis recomendable o la seguridad.
NR: no recomendado.

*Indicados cuando no puedan utilizarse los fármacos de 1ª elección. No hay datos acerca de: etravirina, rilpivirina ni
** Contraindicados con cobicistat.
1 Si el alelo HLA-B*5701 es negativo, aunque con pequeño riesgo de
2 Debe considerarse especialmente para el tratamiento en pacientes con coinfección por VHB. Además, datos publicados sugieren que es un fármaco eficaz y probablemente
3 ZDV es el fármaco antirretroviral con el que más experiencia se tiene en el embarazo. El hecho de considerarlo como alternativo se debe únicamente a ser un tratamiento subóptimo para la
5 Datos
6 Siempre que la CVP sea <100.000 cop/mL y la cifra de linfocitos CD4+ >200/μL (véanse las recomendaciones generales del TAR de inicio).
7 Categoría D, potencialmente teratógeno, aunque datos observacionales sugieren que no hay mayor riesgo de malformaciones congénitas. No es de elección en las 8 primeras semanas de gestación. Si la mujer está tomando ya EFV cuando queda embarazada, es posible continuar su
8 Hay evidencia de que ATV/r es igual de efectivo que DRV/r en embrazadas y se tolera
9 Cuando se acompaña de TDF en lugar de ZDV, es imprescindible utilizar la dosis de 400/100 mg en el segundo y tercer trimestre, dada la interacción existente entre TDF y ATV y las especificidades farmacocinéticas de la gestación.
10 Aunque algunos estudios sugieren que la dosis de 800/100 QD podría ser suficiente en mujeres sin mutaciones de resistencia, parece más seguro utilizar la dosis BID. En mujeres con CVP indetectable con la dosis 800/100 QD puede continuarse con esta dosis con vigilancia estrecha de la
11 No se recomienda con TDF/FTC.
Bibliografía:
Henn A, Flateau C, Gallien S. Primary HIV infection: clinical presentation, testing, and
treatment. Curr Infect Dis Rep 2017;19:37.Robb ML, Eller LA, Kibuuka H, et al. Prospective study of acute HIV-1 infection in adults
in east Africa and Thailand. N Engl J Med 2016; 374:2120-30.Lama JR, Ignacio RAB, Alfaro R, Ríos J, Cartagena JG, Valdez R, Bain C, et al. Clinical and immunologic outcomes after immediate or deferred antiretroviral therapy initiation during primary human immunodeficiency virus infection: The Sabes Randomized Clinical Study. Clin Infect Dis 2021; 72:1042-50.
Colby DJ, Trautmann L, Pinyakorn S, et al. Rapid HIV RNA rebound after antiretroviral
treatment interruption in persons durably supressed in Fiebig I acute HIV infection. Nat
Med 2018; 24:923-6.Saez-Cirion A, Bacchus C, Hocqueloux L, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS pathogens 2013; 9:e1003211.
Cheret A, Nembot G, Melard A, et al. Intensive five-drug antiretroviral therapy regimen versus standard triple-drug therapy during primary HIV-1 infection (OPTIPRIM-ANRS 147): a randomised, open-label, phase 3 trial. Lancet Infect Dis 2015; 15:387-96
Ambrosioni J, Petit E, Liegeon G, Laguno M, Miró JM. Primary HIV-1 infection in users of pre-exposure prophylaxis. Lancet HIV 2021; 8:e166-e174.
de Mendoza C, Cabezas T, Caballero E, et al. HIV type 2 epidemic in Spain: challenges and missing AIDS 2017; 31:1353-64.
Tzou PL, Descamps D, Rhee SY, et al. Expanded spectrum of antiretroviral-selected mutations in HIV-2. J Infect Dis 2020; 221:1962-72.
Raugi D, Ba S, Cisse O, et al. Long-term experience and outcomes of programmatic antiretroviral therapy for HIV-2 infection in Senegal, West Africa. Clin Infect Dis 2021; 72:369-78.
Requena S, Lozano A, Caballero E, et Clinical experience with integrase inhibitors in HIV-2 infected individuals in Spain. J Antimicrob Chemother 2019; 74: 1357-62.
Le Hingrat Q, Collin G, Ghosn J, et Ibalizumab shows in vitro activity against group A and group B HIV-2 clinical isolates. Reviews in Antiviral Therapies & Infectious Diseases 7, 2020. Presented at European Meeting on HIV and Hepatitis 2020. Abstract 75. Disponible en: https://academicmedicaleducation.com/meeting/european-meeting- hiv-hepatitis-2020/abstract/ibalizumab-shows-vitro-activity-against-0.
Recommendations for the use of antiretroviral drugs in pregnant women with HIV- 1 infection and interventions to reduce perinatal HIV transmission in the United States. Departament of health and human services (HHS) Panel on treatment of pregnant women with HIV infection and prevention of perinatal December 30,2021. Accesible en https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/Perinatal_GL.pdf
Mandelbrot L, Tubiana R, Le Chenadec J, et al. No perinatal HIV-1 transmission from women with effective antiretroviral therapy starting before Clin Infect Dis 2015; 61:1715-25.
Siemieniuk RA, Foroutan F, Mirza R, et al. Antiretroviral therapy for pregnant women living with HIV or hepatitis B: a systematic review and meta-analysis. BMJ open 2017; 7:e019022.
Zash R, Holmes L, Diseko M, et al Neural-tube defects and antiretroviral treatment regimens in N Engl J Med 2019; 381:827-40.
Lockman S, Brummel SS, Ziemba L, et IMPAACT 2010/VESTED Study Team and Investigators. Efficacy and safety of dolutegravir with emtricitabine and tenofovir alafenamide fumarate or tenofovir disoproxil fumarate, and efavirenz, emtricitabine, and tenofovir disoproxil fumarate HIV antiretroviral therapy regimens started in pregnancy (IMPAACT 2010/VESTED): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet 2021; Apr 3;397(10281):1276-1292.
Lawn SD, Torok ME, Wood R. Optimum time to start antiretroviral therapy during HIV- associated opportunistic infections. Curr Opin Infect Dis 2011; 24:34-42.
Zolopa A, Andersen J, Powderly W, et al. Early antiretroviral therapy reduces AIDS progression/death in individuals with acute opportunistic infections: a multicenter randomized strategy PLoS One 2009; 4:e5575.
Schäfer G, Hoffmann C, Arasteh K, et al. Immediate versus deferred antiretroviral therapy in HIV-infected patients presenting with acute AIDS-defining events (toxoplasmosis, Pneumocystis jirovecii-pneumonia): a prospective, randomized, open-label multicenter study (IDEAL-study). AIDS Res Ther 2019; 16:34.
Boulware DR, Meya DB, Muzoora C, et Timing of antiretroviral therapy after diagnosis of cryptococcal meningitis. N Engl J Med 2014; 370:2487-98.
Eshun-Wilson I, Okwen MP, Richardson M, Bicanic T. Early versus delayed antiretroviral treatment in HIV-positive people with cryptococcal meningitis. Cochrane Database Syst Rev 2018; 7:CD009012.
Rivero A, Pulido F (coordinadores). Recomendaciones de GeSIDA sobre el tratamiento de la tuberculosis en adultos infectados por el virus de la inmunodeficiencia humana (actualización mayo 2018). http://www.GeSIDA-seimc.org/wp-content/uploads/2018/08/ pdf (Consultado 12.10.2021).
Atwine D, Baudin E, Gelé T, Muyindike W, Mworozi K, Kyohairwe R, et al; ANRS 12292 Rifavirenz study group. Effect of high-dose rifampicin on efavirenz pharmacokinetics: drug-drug interaction randomized trial. J Antimicrob Chemother 2020; 75:1250-58.
Grinsztejn B, De Castro N, Arnold V, et al. Raltegravir for the treatment of patients co- infected with HIV and tuberculosis (ANRS 12 180 Reflate TB): a multicentre, phase 2, non-comparative, open-label, randomised Lancet Infect Dis 2014; 14:459-67.
Dooley KE, Kaplan R, Mwelase N, et al. Dolutegravir-based antiretroviral therapy for patients co-infected with tuberculosis and HIV: a multicenter, noncomparative, open-label, randomized Clin Infect Dis 2020; 70:549-56.
De Castro N, Marcy O, Chazallon C et al; ANRS 12300 Reflate TB2 study group. Standard dose raltegravir or efavirenz-based antiretroviral treatment for patients co- infected with HIV and tuberculosis (ANRS 12 300 Reflate TB 2): an open-label, non- inferiority, randomised, phase 3 Lancet Infect Dis 2021; 21:813-22.
Ford SL, Lou Y, Lewis N, et al. Effect of rifabutin on the pharmacokinetics of oral cabotegravir in healthy Antivir Ther 2019; 24:301-8.
Meintjes G, Stek C, Blumenthal L, et al. Prednisone for the prevention of paradoxical tuberculosis associated N Engl J Med 2018; 379:1915-25.
Ekrikpo UE, Kengne AP, Bello AK, et al. Chronic kidney disease in the global adult HIV- infected population: A systematic review and meta-analysis. PLoS One 2018; 13:e0195443.
Cristelli MP, Cofán F, Rico N, et al. Estimation of renal function by CKD-EPI versus MDRD in a cohort of HIV-infected patients: a cross-sectional BMC Nephrol 2017; 18:58.
Panel de expertos de Documento de consenso de GeSIDA para la evaluación y el tratamiento de las enfermedades renales en pacientes con infección por el virus de la inmunodeficiencia humana. (Actualización Marzo 2020). http://gesida-seimc.org/wp- content/uploads/2020/07/TAR_GUIA_GESIDA_2020_COMPLETA_Julio.pdf (Consultado el 20-08-2021)
Magee M, Slater J, Mannino F, et al. Effect of renal and hepatic impairment on the pharmacokinetics of temsavir, the active moiety of fostemsavir. J Clin Pharmacol 2021; 61:939-53.
Parasrampuria R, Ford SL, Lou Y, et al. A Phase I study to evaluate the pharmacokinetics and safety of cabotegravir in adults with severe renal impairment and healthy matched control Clin Pharmacol Drug Dev 2019; 8:674-81.
Pozniak A, Arribas JR, Gathe J, et Switching to tenofovir alafenamide, coformulated with elvitegravir, cobicistat, and emtricitabine, in HIV-infected patients with renal impairment: 48-week results from a single-arm, multicenter, open-label phase 3 study. J Acquir Immune Defic Syndr 2016; 71:530-7.
Guía AEEH-SEIMC de tratamiento de la infección por virus de la Hepatitis C 2018. Accesible en: https://seimc.org/contenidos/gruposdeestudio/gehep/dcientificos/documentos/gehep-seimc_AEEH-dc-2018-HepatitisC.pdf (accedido el 30/11/2019).
Maurice JB, Patel A, Scott AJ, et al. Prevalence and risk factors of nonalcoholic fatty liver disease in HIV-monoinfection. AIDS 2017; 31:1621-32.
Cervo A, Milic J, Mazzola G, et Prevalence, predictors, and severity of lean nonalcoholic fatty liver disease in patients living with Human immunodeficiency virus. Clin Infect Dis 2020; 71:e694-e701. doi: 10.1093/cid/ciaa430.
Pineda JA, Garcia-Garcia JA, Aguilar-Guisado M, et Clinical progression of hepatitis C virus-related chronic liver disease in human immunodeficiency virus-infected patients undergoing highly active antiretroviral therapy. Hepatology 2007; 46:622-30.
Price H, Dunn D, Pillay D, et al. Suppression of HBV by tenofovir in HBV/HIV coinfected patients: a systematic review and meta-analysis. PLoS One 2013; 8:e68152
Gallant J, Brunetta J, Crofoot G, et Efficacy and safety of switching to a single-tablet regimen of elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide in HIV-1/hepatitis B-coinfected adults. J Acquir Immune Defic Syndr 2016; 73:294-8.
Chan HL, Fung S, Seto WK, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of HBeAg-positive chronic hepatitis B virus infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol 2016; 1:185-95.
Buti M, Gane E, Seto WK, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of patients with HBeAg-negative chronic hepatitis B virus infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol 2016; 1:196-206.
Kim HN, Newcomb CW, Carbonari DM, et al; North American AIDS Cohort Collaboration on Research, Design of IeDEA. Risk of HCC With Hepatitis B Viremia Among HIV/HBV- Coinfected Persons in North America. Hepatology 2021. doi: 10.1002/hep.31839. Epub ahead of PMID: 33780007
Macias J, Mancebo M, Merino D, et Changes in liver steatosis after switching from efavirenz to raltegravir among human immunodeciency virus-infected patients with nonalcoholic fattly liver disease. Clin Infect Dis 2017; 65:1012-9.
Song IH, Borland J, Savina PM, et al. Pharmacokinetics of single-dose dolutegravir in HIV-seronegative subjects with moderate hepatic impairment compared to healthy matched controls. Clinical Pharmacol Drug Dev 2013; 2:342-8
Merli M, Galli L, Marinaro Pharmacokinetics of dolutegravir and rilpivirine in combination with simeprevir and sofosbuvir in HIV/hepatitis C virus-coinfected patients with liver cirrhosis. J Antimicrob Chemother 2017; 72:812-5.
Hernandez-Novoa B, Moreno A, Perez-Elias MJ, et al. Raltegravir pharmacokinetics in HIV/ HCV-coinfected patients with advanced liver cirrhosis (Child-Pugh C). J Antimicrob Chemother 2014; 69: 471-5.
HEP drug interaction University of Liverpool. http://www.hep-druginteractions. org/ (Consultada el 01.09.2021)
Panel de expertos de Guía de práctica clínica sobre los Tumores No Definitorios de SIDA e Infección por el VIH (actualización marzo 2019). Accesible en: http:// GeSIDA-seimc.org/wp-content/uploads/2019/05/GeSIDA_DC_TumoresNoDefinitorios_ Marzo_2019_14_05_19.pdf. (Consultada el 1.10.2021).
Recomendaciones de GeSIDA/PETHEMA sobre el diagnóstico y tratamiento de los linfomas en pacientes infectados por el virus de la inmunodeficiencia humana. Accesible en: http://GeSIDA-seimc.org/wp-content/uploads/2017/08/GeSIDA_Manual_Linfomas_ pdf (Consultada el 1.10.2021).
Guía de práctica clínica de la infección por herpes virus humano tipo 8 en la población con infección por VIH Versión 1.0. – Enero de 2021. Accesible en: https://gesida-seimc. org/wp-content/uploads/2021/03/2021_Guia_GeSIDA.pdf (Consultada el 10.2021).
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). Cancer in people with Version 2.2021-April 26, 2021. Accesible en: https://www.nccn. org/login?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/hiv.pdf (Consultada el 1.10.2021).
Torres HA, Rallapalli V, Saxena A, et al. Efficacy and safety of antiretrovirals in HIV- infected patients with cancer. Clin Microbiol Infect 2014; 20:O672-9.
8. COSTE COMPARATIVO DE LAS DIFERENTES COMBINACIONES DE FÁRMACOS ANTIRRETROVIRALES
El TAR ha reducido la mortalidad relacionada con el SIDA y ha mejorado la calidad de vida de los pacientes. Sin embargo, su coste es elevado y, en un entorno donde los recursos son limitados, es necesario gestionar adecuadamente el presupuesto. Los precios del TAR se han reducido en los últimos años con la comercialización de un número cada vez mayor de genéricos. Sin embargo, pueden existir diferencias de más de 200 euros mensuales en el precio de las terapias de inicio. Una evaluación farmacoeconómica con objeto de determinar el posicionamiento de nuevas estrategias o de nuevos medicamentos debe contemplar no solamente el coste, sino también la eficacia (ensayos clínicos) o la efectividad (práctica clínica habitual) de forma conjunta. Por este motivo, en los últimos años se ha publicado en relación con estas guías, pero posterior en el tiempo, un estudio farmacoeconómico1 en el que se realiza una evaluación de costes y eficiencia (coste/eficacia) mediante construcción de árboles de decisión a partir de las pautas preferentes y alternativas recomendadas. El precio que el Sistema Nacional de Salud paga por un fármaco se obtiene a partir del precio de venta laboratorio (PVL) con la deducción obligatoria del 7,5% sobre el precio de compra de medicamentos no genéricos y no afectados por el sistema de precios de referencia (Real Decreto-ley 8/2010, de 20 de mayo: Medidas extraordinarias para la reducción del déficit público). En el caso de que hubieran transcurrido 10 años desde la fecha de inicio de financiación con fondos públicos (11 años en el caso de haber sido autorizada una nueva indicación), la deducción sería del 15%, salvo en los medicamentos que cuenten con protección de patente de producto en todos los estados miembros de la Unión Europea (Real Decreto-ley 9/2011, de 19 de agosto). A esta cifra habría que sumar el 4% de IVA. Además, cabe considerar que pueden existir variaciones entre los precios finales de adquisición entre distintas comunidades autónomas e, incluso, entre distintos hospitales en una misma comunidad. Por ello, es importante que cada centro utilice sus propios precios para obtener datos de eficiencia. A tal efecto, el estudio mencionado1 ofrece una aplicación informática en la que, introduciendo los precios que cada hospital paga, se obtiene el posicionamiento de las distintas pautas de inicio. La aplicación puede descargarse de forma gratuita en la web de GeSIDA, en “Guías clínicas”, en el apartado http://GeSIDA-seimc.org/category/ guias- clinicas/antirretroviral-vigentes/ (Aplicación cálculo coste y eficacia). Otra consideración a tener en cuenta es que los estudios farmacoeconómicos probablemente subestimen la efectividad del TAR, dado que habitualmente no incluyen la reducción en el riesgo de transmisión de la enfermedad de los pacientes tratados. Este hecho puede tener un impacto económico importante.
Recomendaciones:
- Se recomienda incluir criterios de coste-efectividad en la toma de decisiones sobre el TAR de inicio (A-III).
Bibliografía:
Pérez-Molina JA, Martínez E, Blasco AJ, et al. Analysis of the costs and cost-effectiveness of the guidelines recommended by the 2018 GeSIDA/Spanish National AIDS Plan for initial antiretroviral therapy in HIV-infected adults. Enferm Infecc Microbiol Clin. 2019;37:151-9.
9. DECLARACIÓN DE TRANSPARENCIA (CONFLICTOS DE INTERÉS)
Antonio Antela ha efectuado labores de consultoría para los laboratorios Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare; ha recibido honorarios como investigador en ensayos clínicos de Gilead Sciences, Janssen Cilag, y ViiV Healthcare; y ha recibido compensación económica por charlas o actividades de formación de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Jose Ramón Arribas ha efectuado labores de consultoría para los laboratorios Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme, ViiV Healthcare, Teva, Alexa y Serono; ha disfrutado de becas para investigación clínica de ViiV Healthcare y Gilead Sciences;
y ha recibido compensación económica por charlas de Merck Sharp & Dohme y ViiV Healthcare.
Juan Ambrosioni ha recibido financiación para investigación de Gilead Sciences y ViiV Healthcare. Ha participado en Abvisory Boards para Gilead Sciences, ViiV Healthcare y Janssen pharmaceuticals. DSMB boards para HIPRA y Grifols. Compensación económica por charlas de Gilead Sciences, ViiV Healthcare y Janssen pharmaceuticals.
Victor Asensi ha efectuado labores de consultoría para AbbVie, Gilead Sciences, Janssen Cilag, Janssen Therapeutics, Merck Sharp & Dohme y ViiV Healthcare. Ha recibido becas de investigación de ViiV Healthcare, y honorarios por charlas de AbbVie,
Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Enrique Bernal ha efectuado labores de consultoría para AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag, Janssen Therapeutics, Merck Sharp & Dohme y ViiV Healthcare. Ha recibido becas de investigación de Gilead Sciences, y honorarios por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
José R. Blanco ha efectuado labores de consultoría para AbbVie, Bristol-MyersSquibb, Gilead Sciences, Janssen Cilag, Janssen Therapeutics, Merck Sharp & Dohme y ViiV Healthcare. Ha recibido becas de investigación de Bristol-Myers Squibb y Gilead Sciences, y honorarios por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Alfonso Cabello ha efectuado labores de consultoría para Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme, ViiV Healthcare; ha disfrutado de becas para investigación clínica de ViiV Healthcare y ha recibido compensación económica por charlas de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare; ha recibido ayudas para la asistencia a congresos de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Jose Luis Casado ha efectuado labores de consultoría para Gilead Sciences, Janssen Cilag y ViiV Healthcare. Ha recibido ayudas para la investigación y honorarios por conferencias de Janssen Cilag, Gilead y ViiV Healthcare.
Carmen de Mendoza ha recibido compensación económica por charlas de Abbott Laboratorios y Roche. Su institución ha recibido becas para estudios y proyectos de investigación en los que es investigador principal.
Carlos Dueñas Ha efectuado labores de consultoría para Gilead Sciences, Janssen Cilag, Janssen Cilag y ViiV Healthcare. Ha recibido honorarios por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV
Healthcare.
Ana González-Cordón ha recibido compensación económica por charlas de AbbVie, Boehringer, Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare; ha efectuado labores de consultoría para ViiV Healthcare; ha recibido ayudas para la asistencia a congresos de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Juan González-García ha efectuado labores de consultoría para ViiV, Gilead Sciences, Janssen and Merck Sharp & Dohme. Ha recibido ayudas para investigación y compensaciones económicas por charlas de Gilead Sciences, Janssen, Merck Sharp & Dohme and ViiV Healthcare. Ha recibido igualmente ayudas económicas para asistir a reuniones científicas de Gilead Sciences and Janssen.
Carmen Hidalgo ha efectuado labores de consultoría para Angellini, ViiV, Gilead Sciences, Janssen, y Merck Sharp & Dohme. Ha recibido ayudas para investigación de Gilead Sciences, y Angellini, y compensaciones económicas por charlas Gliead Sciences, Janssen, Merck Sharp & Dohme ViiV Healthcare. Ha recibido igualmente ayudas económicas para asistir a reuniones científicas de Gilead Sciences, ViiV, Janssen Merck
Sharp & Dohme.
Juan Emilio Losa ha efectuado labores de consultoría para ViiV Healthcare y ha recibido honorarios por charlas y ayudas para asistencia a congresos de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Josep Mallolas ha efectuado labores de consultoría para Gilead Sciences, Janssen Cilag, MSD y ViiV Healthcare. Ha recibido ayudas para la investigación y honorarios por conferencias de Janssen Cilag, Gilead, MSD y ViiV Healthcare.
Ana Mariño ha recibido ayudas para asistencia a congresos y reuniones de los laboratorios Gilead Sciences, Janssen Cilag y ViiV Healthcare.
Esteban Martínez ha recibido honorarios por realización de actividades educativas médicas o participación en consejos asesores de las siguientes compañías: Gilead, Janssen, MSD y ViiV. Su institución ha recibido becas para estudios de investigación clínica de MSD y ViiV.
Mar Masiá ha recibido honorarios por realización de actividades educativas médicas, participación en consejos asesores o becas para asistencia a congresos de Janssen, Merck Sharp & Dohme, ViiV Healthcare y Pfizer.
Celia Miralles ha recibido honorarios por asesoría y charlas con fines educativos de Gilead Sciences, Merck Sharp & Dohme y ViiV Healtcare, y becas para asistencia a congresos y reuniones de Gilead Sciences y Merck Sharp & Dohme.
Rocío Montejano ha efectuado labores de consultoría para los laboratorios Gilead Sciences, Merck Sharp & Dohme y ViiV Healthcare; y ha recibido compensación económica por charlas de Gilead Sciences, ViiV Healthcare, Jannsen Cilag y Thera Technologies.
Marta Montero ha efectuado labores de consultoría para los laboratorios Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare; ha recibido compensación económica por charlas de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV
Healthcare; y ha recibido ayudas para investigación de Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Mª Luisa Montes ha efectuado labores de consultoría para los laboratorios Janssen Cilag y Gilead Sciences: Ha disfrutado de becas para la investigación clínica de ViiV,Merck-Sharp & Dome, Abbvie e Intercept. Fondo de Investigaciones Sanitarias (FIS)del Instituto de Salud Carlos III (Madrid), Fundación para la Investigación y Prevención del SIDA en España (FIPSE, Madrid), Ministerio de Sanidad, Servicios Sociales e Igualdad (MSSSI, Madrid). Ha recibido compensaciones económicas por charlas de Abbvie, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag,Merck-Sharp & Dome y Roche.
Rosario Palacios ha efectuado labores de consultoría para los laboratorios Bristol-Myers Squibb, Gilead Sciences, Merck Sharp & Dohme y ViiV Healthcare; ha recibido compensación económica por charlas de Bristol-Myers Squibb, Gilead Sciences, Janssen
Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Roger Paredes ha efectuado labores de consultoría para Merck Sharp & Dohme, Gilead Sciences, Lilly, Theratecnologies, AELIX Therapeutics y ViiV Healthcare; ha disfrutado de ayudas para investigación clínica de Merck Sharp & Dohme, Gilead Sciences y ViiV Healthcare; y ha recibido ayudas para asistencia a congresos de Gilead Sciences, Janssen Cilag y Merck Sharp & Dohme.
José A. Pérez Molina ha efectuado labores de consultoría para Gilead Sciences, Merck Sharp & Dohme y ViiV Healthcare. Ha recibido becas para investigación de Janssen y MSD y ha recibido honorarios por ponencias científicas de Merck Sharp & Dohme,
Theratecnologies y ViiV Healthcare.
José Antonio Pineda ha efectuado labores de consultoría para los laboratorios Abbvie, Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck Sharp & Dohme y ViiV Healthcare; ha disfrutado de becas para investigación clínica de Abbvie, Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck Sharp & Dohme y ViiV Healthcare y ha recibido compensación económica por charlas de Abbvie, Bristol-Myers Squibb, Gilead
Sciences, Janssen-Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Daniel Podzamczer ha recibido honorarios institucionales para financiar investigaciones de parte de Viiv, Gilead y Merck Sharp & Dohme y personales por asesoramiento cientifico y/o charlas de Janssen, Viiv, Gilead y Merck Sharp & Dohme.
Rosa Polo declara no haber recibido ninguna ayuda ni subvención relacionada con este documento.
Eva Poveda ha recibido ayudas para asistencias a congresos de ViiV Healthcare, ha realizado labores de consultoría y participado en actividades de educación biomédica para Theratechnologies y Merck Sharp & Dohme.
Federico Pulido ha efectuado labores de consultoría para los laboratorios Gilead Sciences, Janssen, Merck Sharp & Dohme, Thera y ViiV Healthcare; ha recibido compensación económica por charlas de Gilead Sciences, Janssen Cilag, Merck Sharp
& Dohme y ViiV Healthcare.
Antonio Rivero ha efectuado labores de consultoría para los laboratorios Abbvie, Bristol-Myers Squibb, Gilead Sciences, Merck Sharp & Dohme y ViiV Healthcare; ha disfrutado de becas para investigación clínica de Abbvie, Bristol-Myers Squibb, Gilead Sciences, Merck Sharp & Dohme y ViiV Healthcare; ha recibido compensación económica por charlas de Abbvie, Bristol-Myers Squibb, Gilead Sciences, Merck Sharp & Dohme y ViiV Healthcare.
Jesús Santos ha recibido compensación económica por charlas de Gilead Sciences, Janssen Cilag, MerckSharp; Dohme y ViiV Healthcare; ha efectuado labores de consultoría para ViiV Healthcare; ha recibido ayudas para la asistencia a congresos de Gilead Sciences.
Jesús Sanz Sanz ha efectuado labores de consultoría para los laboratorios Abbvie, Boehringer Ingelheim Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare; ha recibido compensación económica por charlas de Abbvie, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare, y ha recibido pagos por desarrollo de presentaciones educacionales de ViiV Healthcare.
Sergio Serrano ha efectuado labores de consultoría para Merck Sharp & Dohme y Gilead Sciences, ha disfrutado de becas para investigación clínica de Merck Sharp & Dohme y Gilead Sciences, ha recibido pagos por desarrollo de presentaciones docentes de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y Janssen y ha recibido becas para la asistencia a congresos y reuniones de ViiV Healthcare y Gilead Sciences.
Inés Suárez ha efectuado labores de consultoría para los laboratorios ViiV Healthcare y Merck Sharp & Dohme, ha recibido compensación económica por presentaciones docentes de GILEAD, Viiv Healthcare, Merck Sharp & Dohme, Janssen y Bristol-Myers Squibb, y ha recibido becas para la asistencia a congresos y reuniones de ViiV Healthcare, Merck Sharp & Dohme, Janssen y GILEAD.
Miguel Torralba ha efectuado labores de consultoría para los laboratorios Gilead Sciences, Merck Sharp & Dohme y ViiV Healthcare; ha recibido compensación económica por presentaciones para Abbvie, Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Jesús Troya ha recibido compensación económica por charlas formativas de Gilead Sciences, Janssen Cilag y Merck Sharp & Dohme.
Montserrat Tuset ha disfrutado de becas para investigación clínica de Janssen, Gilead Sciences y ViiV Healthcare y ha recibido compensación económica por charlas de Gilead Sciences, Janssen, MerckSharp & Dohme, ViiV Healthcare y Theratechnologies.
María Velasco ha recibido honorarios y/o ayudas para actividades con fines docentes o ponencias de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Isabel Viciana ha efectuado labores de consultoría para los laboratorios Gilead Sciences; ha recibido compensación económica por charlas de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare. Ha recibido ayudas para asistencias a congresos de Gilead Sciences y ViiV Healthcare.
María Jesús Vivancos ha impartido presentaciones con fines docentes de AbbVie, Gilead Sciences y ViiV Healthcare por las que ha recibido compensación económica y ha asistido de forma esporádica a congresos con la financiación de Gilead, ViiV Healthcare y Janssen Cilag.
Miguel Angel von Wichmann ha efectuado labores de consultoría para los laboratorios Abbvie, Bristol-Myers Squibb, Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare; ha recibido compensación económica por charlas de Abbvie, Bristol- Myers Squibb, Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ha recibido pagos por desarrollo de presenta.