Guía de práctica clínica de la infección por herpes virus humano tipo 8 en la población con infección por el VIH
Octubre 2021
Notas de la Versión:
Octubre 2020
LISTADO DE ABREVIATURAS
ADN |
Ácido desoxirribonucléico |
ARN |
Ácido ribonucléico |
bFGF |
Factor de crecimiento derivado de fibroblastos |
CHOP |
Ciclofosfamida, Adriamicina, Vincristina, Prednisona |
DA-EPOCH |
EPOCH con dosis ajustadas |
EC |
Enfermedad de Castleman |
ECM |
Enfermedad de Castleman multicéntrica |
EPOCH |
Etopósido, Prednisona, Vincristina, Ciclofosfamida, Adriamicina |
FDA |
Food and Drug Administration |
G-CSF |
Factor estimulantes de los granulocitos |
HSH |
Hombres que tienen sexo con hombres |
HVSK |
Herpes virus asociado al sarcoma de Kaposi |
IDSA |
Infectious Diseases Society of America |
Ig |
Inmunoglobulina |
IL |
Interleukina |
IP |
Inhibidor de la proteasa |
KICS |
Síndrome inflamatorio inducido por citokinas |
LANA-1 |
Antígeno nuclear asociado a latencia |
LNH |
Linfoma no Hodgkin |
LPC |
Linfoma primario de cavidades |
OMS |
Organización mundial de la salud |
PCR |
Protein Chain Reaction |
PDGF |
Factor de crecimiento derivado de las plaquetas |
PET-TC |
Tomografía computarizada de emisión de positrones |
RC |
Respuesta completa |
SG |
Supervivencia global |
SIRI |
Síndrome inflamatorio de reconstitución inmune |
SK |
Sarcoma de Kaposi |
SRI |
Síndrome de reconstitución inmune |
TAR |
Tratamiento antirretroviral |
TARGA |
Tratamiento antirretroviral de gran actividad |
TC |
Tomografía computarizada |
VEB Virus de Epstein Barr
v-FLIP Proteína inhibidora viral de FLICE
VHH-8 Virus herpes humano tipo 8
VIH Virus de la inmunodeficiencia humana
1. INTRODUCCIÓN
El Sarcoma de Kaposi (SK) es un tumor mesenquimal originado por el virus herpes humano tipo-8 (VHH-8) que, de forma global, continúa siendo el más frecuente en la población con infección por VIH. Tras la utilización del tratamiento antirretroviral (TAR), su frecuencia ha disminuido y su pronóstico ha mejorado. Sin embargo, el riesgo de desarrollar SK continúa siendo más elevado en las personas con VIH y recuento normal de linfocitos CD4+ que en la población general. Los mecanismos inmunopatológicos por los que hasta un 15% de los pacientes con carga viral indetectable y linfocitos CD4+ > 300 por mm 3 pueden desarrollar enfermedad progresiva permanecen todavía desconocidos 15 .
Objetivo y alcance
El objetivo de esta guía es familiarizar a los profesionales que atienden a los pacientes con infección por el VIH con la presentación clínica, diagnóstico y tratamiento del SK y otras enfermedades relacionadas con el VHH-8 en la era del TAR eficaz.
Metodología
Se incluye en esta guía información sobre el SK, la enfermedad de Castleman multicéntrica (ECM), el linfoma primario de cavidades (LPC) y el síndrome inflamatorio asociado a citoquinas (KICS). Dada la poca información proveniente de ensayos clínicos en cuanto a tratamiento antineoplásico específico e inmunoterapia, se han revisado fundamentalmente los estudios observacionales, las revisiones sistemáticas y las guías nacionales e internacionales 6 . Cada apartado de esta Guía ha sido realizado por un redactor y revisado por un revisor. Tanto los redactores como los revisores han sido designados por la Junta Directiva de GeSIDA después de valorar el curriculum vitae de las personas que se ofrecieron para la redacción del documento. Dos de los miembros del panel han actuado como coordinadores cuyo cometido, además de redactar alguno de los apartados, ha sido ensamblar los distintos apartados y encargarse de la redacción y edición final del mismo. El documento final ha sido consensuado por todo el Panel. En esta guía la fuerza de la recomendación y gradación de las pruebas que la sustentan se basan en una modificación de los criterios de la Infectious Diseases Society of America (IDSA) . Según estos criterios cada recomendación debe ofrecerse siempre (A), en general (B) u opcionalmente
(C) y ello basado en la calidad de los datos obtenidos a partir de uno o más ensayos clínicos aleatorizados con resultados clínicos o de laboratorio (I), de uno o más ensayos no aleatorizados o datos observacionales de cohortes (II) o de la opinión de expertos (III) Tabla 1
Tablas:

Bibliografía:
Dalla Pria A, Pinato DJ, Bracchi M and Bower Recent advances in HIV-associated Kaposi sarcoma [version 1; peer review: 2 approved] F1000Research 2019, 8(F1000 Faculty Rev):970 ( https://doi.o rg/10.12688/ f1000research.17401.1 )
Cañas-Ruano E, Martín-Castillo M, Raventós B, Burgos J, Curran A, Navarro J et al. Incidence of malignancy in a Spanish cohort of patients infected by the human immunodeficiency Med Clin (Barc). 2020 Jan 28. pii: S0025-7753(19)30720-1. doi: 10.1016/j.medcli.2019.12.001.
Goncalves PH, Ziegelbauer J, Uldrick TS, Yarchoan Kaposi sarcoma herpesvirus-
Poizot-Martin I, Lions C, Cheret A, Rey D, Duvivier C, Jacomet C et Kaposi sarcoma in people living with HIV: incidence and associated factors in a French cohort between 2010 and 2015. AIDS. 2020; 34(4): 569-577. doi: 10.1097/QAD.0000000000002450.
Etemad SA, Dewan AK. Kaposi Sarcoma Updates. Dermatol Clin. 2019; 4: 505-517. doi: 10.1016/j.det.2019.05.0086. Lebbe C, Garbe C, Stratigos AJ, Harwood C, Peris K, Marmol Vdet al. Diagnosis and treatment of Kaposi's sarcoma: European consensus- based interdisciplinary guideline (EDF/EADO/EORTC). Eur J 2019; 114: 117-127.
Reid E, Suneja G, Ambinder RF, Ard K, Baiocchi R, Barta SK et AIDS-Related Kaposi Sarcoma, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2019 Feb; 17(2):171-189. doi: 10.6004/jnccn.2019.0008.
Abramson Diagnosis and Management of Castleman Disease. J Natl Compr Canc
Volkow-Fernández P, Lome-Maldonado C, Quintero-Buenrostro H, Islas-Muñoz B, Cornejo-Juárez HIV-associated multicentric Castleman disease: a report of 19 cases at an oncology institution. Int J STD AIDS. 2020 (4): 318-325. doi: 10.1177/0956462420905277.
Lurain K, Polizzotto MN, Aleman K, Bhutani M, Wyvill KM, Gonçalves PH et al. Viral, immunologic, and clinical features of primary effusion Blood. 2019; 133: 1753- 1761. doi: 10.1182/blood-2019-01-893339.
Cantos VD, Kalapila AG, Ly Nguyen M, Adamski M, Gunthel Experience with Kaposi Sarcoma Herpesvirus Inflammatory Cytokine Syndrome in a Large Urban HIV Clinic in the United States: Case Series and Literature Review. Open Forum Infect Dis. 2017;4(4): ofx196. doi: 10.1093/ofid/ofx196.
Polizzotto MN, Uldrick TS, Wyvill KM, Aleman K, Marshall V, Wang V et al. Clinical Features and Outcomes of Patients With Symptomatic Kaposi Sarcoma Herpesvirus (KSHV)-associated Inflammation: Prospective Characterization of KSHV Inflammatory Cytokine Syndrome (KICS). Clin Infect 2016; 62: 730-738. doi: 10.1093/cid/civ996
Kish MA. Guide to development of practice guidelines. Clin Infect Dis 2001; 32: 851-
2. HERPES VIRUS HUMANO TIPO 8
Características virológicas
El virus herpes humano tipo 8 (VHH-8) denominado también Herpes virus asociado al sarcoma de Kaposi (HVSK), pertenece a la familia Herpesviridae, subfamilia Gammaherpesvirinae junto al virus de Epstein Barr (VEB) y se incluye en el género Rhadinoviridae.
El VHH-8 es un virus cubierto por una membrana fosfolipídica que rodea una nucleocápside con ADN bicatenario, de simetría icosaédrica y replicación en el núcleo celular. El genoma se encuentra muy conservado entre cepas (>90% de identidad). Sin embargo, existen 2 importantes excepciones, el gen K1 y el gen K15. El gen K1 es muy estable para cada individuo, pero extremadamente variable entre cepas de individuos distintos. Este gen codifica una glucoproteína de membrana relacionada con la familia de receptores de inmunoglobulina, que se expresa durante el ciclo lítico y permite distinguir los 7 subtipos virales del VHH-8 (A, B, C, D, E, F y Z), cada uno de los cuales comprende a su vez varios subclados o subgrupos filogenéticos. Se han relacionado los diferentes subtipos con la distribución geográfica y con la progresión de enfermedad 1 . Del gen K15 se han descrito varios alelos distintos que muestran poca variación interindividual. Por ello, la identificación del K1 junto con el alelo de K15 proporciona una herramienta útil para filiar la trazabilidad en la transmisión del VHH-82 .
El VHH-8 es un virus linfotrópico, que tiene replicación lítica en células de la piel, vasos sanguíneos y órganos. Después de una infección primaria, establece un estado de latencia en linfocitos B y en células del endotelio vascular e intermitentemente experimenta periodos de replicación lítica, utilizando la maquinaria celular del huésped para asegurar la trasmisión del virus y el mantenimiento del santuario de latencia. Varios factores internos y externos, como la inflamación, hipoxia, estrés oxidativo, modificaciones epigenéticas y coinfección por otros virus, pueden favorecer el paso de fase latente a replicación lítica. Entre estos, el estado inmunológico del huésped es el factor predominante en el control de la reactivación lítica3.
Durante la fase de latencia no existe producción de virus y solo se expresan unos pocos genes que codifican proteínas como el antígeno nuclear asociado a latencia (LANA), viral- ciclina, proteína inhibidora viral de FLICE (v-FLIP), kaposina y varios miARN, capaces de promover la supervivencia y proliferación celular, así como contribuir en la oncogénesis2 .
El ADN del virus codifica una serie de proteínas similares a las proteínas celulares importantes en la regulación del ciclo celular, en la diferenciación y en la activación celular, así como en la inhibición de la apoptosis natural. El virus al infectar los linfocitos B, promueve la síntesis y liberación por parte de estas células de IL-6, factor de crecimiento derivado de las plaquetas (PDGF) y factor de crecimiento de fibroblastos (bFGF), que presentan actividad estimulante del crecimiento celular. Además, el virus puede favorecer la expresión celular de integrinas, receptores de membrana para factores de crecimiento y otras proteínas de matriz extracelular (Bcl-2 y moléculas de la familia Bcl-X) que incrementan la supervivencia celular, regulan la apoptosis y favorecen la proliferación vascular4.
Epidemiología. Transimisión.
La transmisión del VHH-8 es fundamentalmente a través de la saliva, sin embargo, a diferencia de lo que ocurre con el VEB, es necesario que exista un contacto repetido, como ocurre en la interacción madre-hijo y en las relaciones sexuales. Así en zonas endémicas como en África subsahariana, la alta prevalencia puede ser debido a la trasmisión no sexual del virus a través de la saliva.
De forma general, es más frecuente en hombres que en mujeres y en áreas no endémicas, es más común en hombres que tienen sexo con hombres (HSH)5 . Se ha detectado ADN del VHH-8 en semen, pero de forma más constante en saliva, lo cual refuerza que el contacto con este fluido es el principal factor de riesgo. Otras vías de trasmisión son la sangre y la donación de órganos. La trasmisión transplacentaria es rara.
Aunque el VHH-8 tiene una distribución mundial, existe una gran diferencia entre diferentes áreas geográficas. El Norte de América, gran parte de Europa y Asia presentan una baja prevalencia (<5%), determinadas áreas del Mediterráneo (Sur de Italia, Alejandría), Europa del Este, Caribe y Oriente Medio tienen una prevalencia intermedia (5%-20%) y África y la Amazonia Brasileña presentan la prevalencia más alta (>50%).
Según el área geográfica predominan unos subtipos y subclados del VHH-8. Los subtipos A y C circulan predominantemente en Europa, Norte América y Norte de Asia, mientras que los subtipos D y E están presentes en Taiwán y poblaciones aborígenes de América del Sur, respectivamente. Los subtipos B y A5 han sido encontrados en África subsahariana y los subtipos F y Z en Uganda y Zambia, respectivamente2.
Patología asociada
La mayor parte de las infecciones producidas por el VHH-8 son subclínicas y pueden pasar inadvertidas. La asociación con enfermedades malignas es significativamente más alta en pacientes VIH positivos, pero pueden ocurrir sin coinfección con VIH, siendo las poblaciones con mayor riesgo aquellas que presentan algún grado de inmunodepresión.
Se ha descrito también una posible implicación en la patogenia de enfermedades crónicas como la diabetes mellitus6.
La infección por el HHV8 se ha asociado a los siguientes procesos clínicos7:
- Sarcoma de Kaposi (SK)8 , con 4 variantes:
- Endémico o africano
- Clásico o esporádico o mediterráneo
- Epidémico o asociado a SIDA
- SK asociado a trasplante910
- Linfoma primario de cavidades (LPC)11
- Enfermedad de Castleman multicéntrica (ECM)
- Linfoma de células B grandes en el contexto de ECM
- Síndrome inflamatorio inducido por citoquinas (KICS)
- Síndrome hemofagocítico, pancitopenia, hepatitis.
- Manifestaciones inespecíficas como fiebre, astenia, linfadenopatía, rash y diarrea.
Ideas claves
- La mayor parte de las infecciones son subclínicas y su asociación con enfermedades malignas es significativamente más alta en pacientes VIH+.
- Se transmite por saliva, relaciones sexuales, transfusión y trasplante de órgano sólido.
- El conocimiento del virus, su epidemiología y mecanismos de transmisión permitirán un diagnóstico precoz.
Bibliografía:
Jary A, Leducq V, desire N, et al. New Kaposi sarcoma-associated herpesvirus variant in men who have sex with men associated with severe J Infect Dis 2020; 222 (8):1320-1328.
Bellocchi MC, Svicher V, Ceccherini-Silbrstein F. HHV-8 genetic diversification and its impacto n severe clinical presentation of associated diseases. J Infect Dis 2020; 222 (8): 1250-1253.
Mui UN, Haley CT, Tyring Viral oncology: molecular biology and pathogenesis. J Clin Med 2017; 6 (12):111.
Yan L, Majerciak V, Zheng Z-M, et Towards better understanding of KSHV life cycle: from transcription and posttranscriptional regulations to pathogenesis. Virol Sin 2019; 34 (2): 135-161.
Liu Z, Fang Q, Zuo J, et al. Global epidemiology of human herpesvirus B in men who sex with men: a systematic review and meta-analysis. J Med Virol 2018; 90 (3): 582-591.
Angius F, Ingianni A, Pompei R. Human Herpesvirus 8 and host-cell interaction: long-lasting physiological modifications, inflammation and related chronic Microorganisms 2020; 8 (3): 388.
Gonçalves PH, Uldrick TS, Yarchoan HIV-associated Kaposi Sarcoma and Related
Cesarman E, Damania B, Krown SE, et Kaposi Sarcoma. Nat Rev Dis Primers 2019; 5 (1): 9.
Pellet Madam R, Hand J, on behalf on the AST Infectious Diseases Community of Human herpesvirus 6, 7, and 8 in solid organ transplantation: Guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin Transplant 2019; 33: e13518.
Cahoon EK, Linet MS, Clarke CA, et Risk of Kaposi sarcoma after solid organ
Lurain K, Polizzotto MN, Aleman K, et Viral, immunologic, and clinical features of
3. SARCOMA DE KAPOSI: CLÍNICA Y DIAGNÓSTICO
3.1. Introducción
3.2. Manifestaciones clínicas
3.3. Diagnóstico
3.4. Estadificación y pronóstico
3.5. Ideas clave
Introducción
El Sarcoma de Kaposi (SK) es un angiosarcoma multifocal, que se asocia a la infección crónica por el virus del herpes humano tipo 8 (VHH-8); hasta la aparición de la epidemia del VIH en los años 80, había sido considerado una neoplasia muy rara. Desde entonces, el incremento relacionado con dicha infección le llevó a ser considerada una enfermedad definitoria de SIDA1 , con predominio en el sexo masculino, pasando a ser denominada en tal caso SK epidémico. Sus células tumorales alargadas en forma de huso tienen origen en células endoteliales y/o mesenquimales y están rodeadas de un infiltrado inflamatorio y espacios neovasculares dependiendo el crecimiento de una señal paracrina adicional1 . Antes de la introducción del tratamiento antirretroviral de gran actividad (TARGA), el riesgo de SK en pacientes infectados por el VIH era muy elevado, especialmente en hombres que tenían sexo con hombres (HSH), llegando a desarrollarlo uno de cada cuatro pacientes2. Si bien su incidencia se ha ido reduciendo, sigue siendo una de las neoplasias definitorias de SIDA más frecuente fundamentalmente en el ya mencionado grupo de HSH. También se ha presentado en menor medida en usuarios a droga por vía parenteral, heterosexuales, hemofílicos y niños3. El tratamiento comienza por la restauración del sistema inmune y control de la infección VIH con TARGA; en determinados pacientes, especialmente muy inmunodeprimidos, el comienzo del TARGA puede desencadenar la aparición o el agravamiento de las lesiones de SK, como expresión de síndrome inflamatorio de reconstitución inmune (SIRI)4 . En casos avanzados con afectación sistémica o cutánea extensa (más de 15-25 lesiones), edema intenso, SK cutáneo refractario a tratamiento local, o rápidamente evolutivo, o SK en el contexto de SIRI o con afectación visceral sintomática, se recomienda tratamiento sistémico con quimioterapia56. En relación al pronóstico, existen diferencias relacionadas con la procedencia u origen de los pacientes, así en países pertenecientes al África subsahariana la evolución suele ser menos favorable que en Occidente; en la actualidad se están investigando nuevos tratamientos basados en la patogenia de la enfermedad con resultados prometedores7.
Manifestaciones clínicas
El SK en pacientes VIH+ suele afectar fundamentalmente a piel, siendo habitualmente la primera manifestación de esta neoplasia. Las lesiones se caracterizan por ser indoloras, no pruriginosas, múltiples y afectar fundamentalmente a extremidades, tronco y cara; su color oscila desde el rosa pálido al rojo vinoso, no desaparecen a la vitropresión, pueden ser planas o sobre-elevadas, e incluso ocasionalmente forman nódulos ulcerados, sangrantes, exofíticos, edematosos y dolorosos, con un tamaño desde milímetros a varios centímetros. En el tronco las placas suelen ser grandes y presentarse en los pliegues con aspecto alargado7.
La afectación visceral en el SK está en torno al 15%, no suele ser una manifestación inicial, y en la actualidad es muy poco frecuente por el uso de antirretrovirales; se puede afectar cualquier órgano, aparato o sistema, pero los más frecuentemente implicados son la cavidad oral, el tracto gastrointestinal y el aparato respiratorio. La afectación mucosa ocurre en el 20% de los pacientes infectados por el VIH, y suele presentarse fundamentalmente en paladar y encías, dando lugar en ocasiones a disfagia y sobreinfección bacteriana8.
La afectación del tracto gastrointestinal, particularmente del estómago, en ausencia de la cutánea, puede constituir la primera manifestación de un SK; en la era pre-TARGA el 40% de los pacientes tenían dichas lesiones, llegando a ser hasta del 80% en las autopsias realizadas; el cuadro clínico abarca diferentes signos y síntomas que incluyen desde sangrado, dolor abdominal, nauseas, vómitos, obstrucción intestinal o diarrea, hasta lesiones asintomáticas9. El tratamiento se plantea en función de la sintomatología.
La afectación pulmonar en un 80-90% de los casos suele estar precedida de afectación cutánea extensa o digestiva, en sujetos severamente inmunodeprimidos, y puede afectarse el parénquima, las vías aéreas, la pleura y/o los ganglios linfáticos intra-torácicos. No hay signos o síntomas patognomónicos del SK pulmonar para distinguirlo de otras entidades nosológicas; suele cursar con disnea, tos seca y a veces, hemoptisis, con o sin fiebre, pudiendo representar una amenaza para la vida del paciente7.
Diagnóstico
El diagnóstico suele ser clínico, pero es recomendable confirmarlo mediante biopsia, puesto que según algunos autores el diagnóstico de visu tiene tan solo un valor predictivo positivo del 80% 10.
La radiografía de tórax constituye la técnica inicial de despistaje para evaluar la afectación pulmonar en el SK; lo más frecuente es un infiltrado retículo-nodular con o sin derrame pleural, aunque pueden aparecer patrones mixtos con áreas de consolidación focal, nódulos solitarios, o coalescentes, e incluso es posible la radiografía de tórax normal. El TAC de tórax de alta resolución habitualmente muestra engrosamiento septal interlobar y peribroncovascular (por infiltración de bandas tumorales continuas alrededor de los bronquios y en el intersticio perivascular), nódulos simétricos bilaterales bien definidos distribuidos alrededor de los bronquios y cisuras, con adenopatías mediastínicas y derrame pleural. Existe correlación entre dichos hallazgos radiológicos y los histológicos propios del SK obtenidos mediante broncoscopia y biopsia transbronquial, de forma que este patrón en pacientes con SIDA es altamente sugerente de dicha neoplasia11. En cuanto al papel del PET/TAC en el SK se ha empleado como estudio de extensión y monitorización de la respuesta al tratamiento, cuyos resultados pueden condicionar la actitud terapéutica12.
En relación al SK gastrointestinal no se recomienda endoscopia de rutina en ausencia de síntomas, y de realizarse las lesiones suelen ser tan características que a menudo no se requiere biopsia13. Dichas lesiones incluyen máculas de aspecto angiodisplásico, nódulos submucosos vascularizados, úlceras, máculo-pápulas, placas de diferentes tamaños, e incluso linitis plástica en el estómago.
El diagnóstico anatomo-patológico del SK se realiza mediante la tinción de hematoxilina eosina que se emplea para evaluar todos los tipos de enfermedad, sus características y el grado. La clasificación incluye los estadios de progresión tumoral, que corresponden a la aparición clínica de las lesiones; sin embargo, existe solapamiento entre los diferentes estadios, y no está claro que estos ocurran de forma secuencial. En el examen histológico de las lesiones cutáneas, se observa en el grosor de la dermis proliferación de células fusiformes con infiltración leucocitaria y angiogénesis, con proliferación de vasos pequeños e irregulares desprovistos de membrana basal y formación de hendiduras vasculares. Existe también extravasación de hematíes, depósito de hemosiderina e infiltrado leucoplasmocitario con glóbulos hialinos intra y extracelulares, y el denominado signo del promontorio14. Es importante realizar el diagnóstico diferencial con la angiomatosis bacilar, originada por infección por Bartonella spp (presencia de bacilos gramnegativos en la tinción con plata), y con diversos tumores con proliferación vascular.
La PCR en la biopsia, con amplificación de las secuencias del ADN del VHH-8, permite establecer el diagnóstico. Los marcadores inmunohistoquímicos son útiles en el diagnóstico diferencial de casos complicados; entre estos marcadores se encuentran el CD34 (marcador vascular)15, la vimentina (marcador mesenquimal)16, y especialmente el antígeno nuclear asociado a latencia (LANA-1) del VHH-817, entre otros.
Por otra parte, se ha relacionado el desarrollo de lesiones de SK, con la detección mediante PCR en tiempo real de ADN del VHH-8 en las células mononucleares de sangre periférica, y con test serológicos que evidencian la presencia de títulos elevados de anticuerpos dirigidos frente a LANA-1 y frente a antígenos líticos (K8.1) del VHH-818.
Estadificación y pronóstico
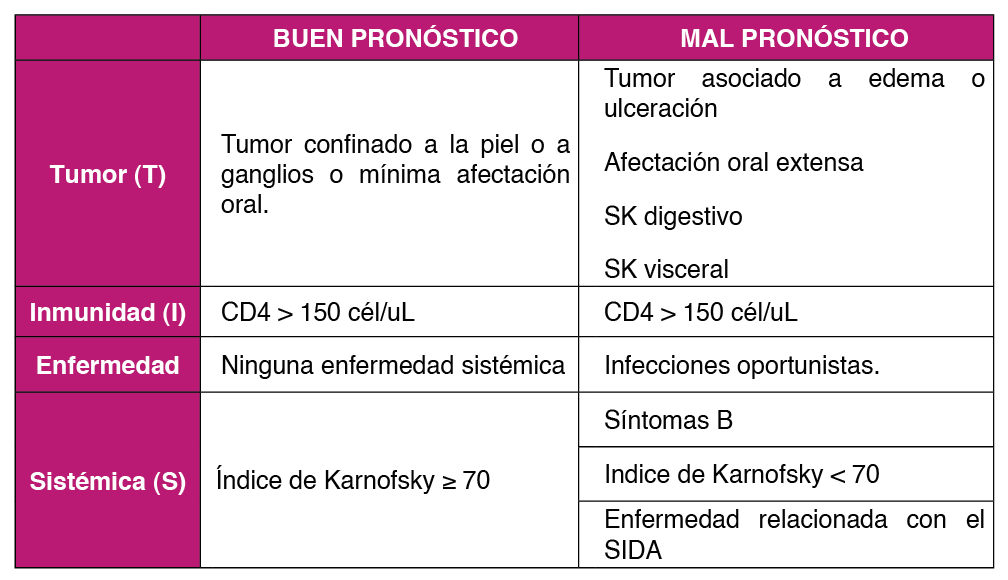
El AIDS Clinical Trials Group Oncology Committee propuso en 1989 y revisó en 1997 una estadificación basada en la extensión (T) y localización de los tumores, estado inmunitario del paciente (I) y compromiso sistémico (S) (infecciones oportunistas, síntomas B como fiebre, pérdida de peso o diarrea persistente, y escala de Karnofsky)1920. A continuación, se detalla la clasificación:
- T0 (buen pronóstico): tumor localizado en la piel y/o nódulos linfáticos y/o en el paladar.
T1 (mal pronóstico): lesiones diseminadas; se considera cuando uno o más de los siguientes ítems están presentes: edema o ulceración en la piel producido por el tumor; SK oral extenso: lesiones que son nodulares y/o en áreas de la boca más allá del paladar (en el suelo de la boca); lesiones de SK en órganos como pulmón, intestino, hígado, etc.
- I0 (buen pronóstico): CD4 ≥ 150 cél/uL
- I1 (mal pronóstico): CD4 < 150 cél/uL.
S0 (buen pronóstico): se considera cuando no hay datos de enfermedad sistémica que incluye: no tener historia de infecciones oportunistas o muguet oral, ni síntomas B en las 2 últimas semanas, ni pérdida de más del 10% de peso sin hacer dieta y Karnosfsky < 70.
S1 (mal pronóstico): enfermedad sistémica presente, Karnofsky < 70, o uno o más de los ítems comentados en S0.
Esta clasificación se realizó antes de la era TARGA y se emplea solo en estudios clínicos19 ( Tabla 1 ).
Desde la introducción del TARGA, el estado inmune (I) ha llegado a ser menos importante y no se tiene en cuenta para categorizar el riesgo2021, de forma que se considera que un paciente tiene buen pronóstico en caso de T0 S0, T1 S0, o T0 S1; y mal pronóstico si T1, S1.
Ideas clave
- El diagnóstico de SK se basa en la clínica, en el estado inmunológico del paciente y, a veces, en los resultados del estudio anatomo-patológico (B-II).
- La radiografía de tórax se recomienda en el despistaje de la afectación pulmonar (B-II).
- No se recomienda de forma rutinaria la realización de endoscopia para descartar SK gastrointestinal (A-II).
- El TARGA constituye una pieza clave en el tratamiento del SK (A-II).
- La afectación cutánea u oral extensa, junto con la presencia de síntomas sistémicos, son los fundamentales condicionantes de mal pronóstico.
Tablas:
SK: sarcoma de Kaposi; VIH: virus de la inmunodeficiencia humana.
Bibliografía:
Fröhlich J, Grundhoff Epigenetic control in Kaposi sarcoma-associated herpesvirus
Martro E, Esteve A, Schulz TF, Sheldon J, Gambus G, Munoz R, et al. Risk factors for human Herpesvirus-8 infection and AIDS-associated Kaposi's Sarcoma among men who have sex with men in a European multicentre Int J Cancer. 2007; 120: 1129-1135.
Silverberg MJ, Lau B, Achenbach CJ, Jing Y, Althoff KN, D'Souza G, et al. Cumulative Incidence of Cancer Among Persons With HIV in North America: A Cohort Study. Ann Intern Med. 2015; 163: 507-18.
Manzardo C, Guardo AC, Letang E, Plana M, Gatell JM, Miro JM. Opportunistic infections and immune reconstitution inflammatory syndrome in HIV-1-infected adults in the combined antiretroviral therapy era: a comprehensive Expert Rev Anti Infect Ther 2015; 13:751-67.
Di Lorenzo G, Kreuter A, di Trolio R, Guarini A, Romano C, Montesarchio V, et Activity and safety of pegylated liposomal doxorubicin as first-line therapy in the treatment of non- visceral classic Kaposi's sarcoma: A multicenter study. J Invest Dermatol. 2008; 128:1578- 1580
Regnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: A systematic review of the J Am Acad Dermatol. 2013; 68: 313-331
Cesarman E, Damania B, Krown SE, Martin J, Bower M, Whitby Kaposi sarcoma. Nat Rev Dis Primers. 2019; 5: 9.
Bower M, Dalla Pria A, Coyle C, Andrews E, Tittle V, Dhoot S, et Prospective stage- stratified approach to AIDS-related Kaposi's sarcoma. J Clin Oncol. 2014; 32: 409.
Danzig JB, Brandt LJ, Reinus JF, Klein Gastrointestinal malignancy in patients with AIDS. Am J Gastroenterol. 1991; 86: 715.
Amerson E, Woodruff CM, Forrestel A, Wenger M, McCalmont T, LeBoit P, et al. Accuracy of clinical suspicion and pathologic diagnosis of Kaposi sarcoma in East
Acquir. Immune. Defic. Syndr. 2016; 71: 295–301.
Gasparetto TD, Marchiori E, Lourenço S, Zanetti G, Vianna AD, Santos AA, et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare 2009; 4: 18.
Mankia SK, Miller RF, Edwards SG, Ramsay A, Lee The response of HIV- associated
Darr U, Renno A, Khan Z, Alkully T, Moslim MA, Kamal S, et al Nawras Endoscopic Appearance of Oropharyngeal and Upper GI Kaposi’s Sarcoma in an Immunocompromised Patient. Case Rep Gastrointest Med. 2017; 2017: 3742684.
Requena C, Alsina M, Morgado-Carrasco D, Cruz J, Sanmartín O, Serra- Guillén C, et al. Sarcoma de Kaposi y angiosarcoma cutáneo: directrices para el diagnóstico y Actas Dermosifiliogr. 2018; 109: 878-887.
Pyakurel P, Pak F, Mwakigonja A R, Kaaya E, Heiden T, Biberfeld P, et al. Lymphatic and vascular origin of Kaposi’s sarcoma spindle cells during tumor development. Int. J. 2006; 119: 1262–1267.
Li Y, Zhong C, LiuD, Yu W, Chen W, Wang Y, et al. Evidence for Kaposi sarcoma originating from mesenchymal stem cell through KSHV-induced mesenchymal-to- endothelial Cancer Res. 2018; 78, 230–245.
Hiatt K M, Nelson A M, Lichy J H, Fanburg-Smith J C. Classic Kaposi sarcoma in the United States over the last two decades: A clinicopathologic and molecular study of 438 non-HIV-related Kaposi sarcoma patients with comparison to HIV-related Kaposi Mod Pathol. 2008; 21: 572-582.
Brown EE, Whitby D, Vitale F, Marshall V, Mbisa G, Gamache C, et al. Virologic, hematologic, and immunologic risk factors for classic Kaposi Cancer 2006; 107: 2282-90.
Krown SE, Metroka C, Wernz Kaposi's sarcoma in the acquired immune deficiency syndrome: a proposal for uniform evaluation, response, and staging criteria. AIDS Clinical Trials Group Oncology Committee. J Clin Oncol. 1989 Sep;7(9):1201-7.
Krown S E, Testa MA, Huang AIDS-related Kaposi's sarcoma: Prospective validation of the AIDS Clinical Trials Group staging classification AIDS Clinical Trials Group Oncology Committee. J Clin Oncol. 1997; 15: 3085-3092
American Cancer Kaposi Sarcoma Stages. April 19, 2018. https://www.cancer. org/cancer/kaposi-sarcoma/detection-diagnosis- staging/staging.htmlwww.cancer.org
4. SARCOMA DE KAPOSI: TRATAMIENTO Y SINDROME DE RECONSTITUCIÓN INMUNE
4.1. Tratamiento anirretroviral
4.2. Terapias tópicas
4.3. Terapias sistémicas
4.4. Tratamiento antiviral frente al VHH-8
4.5. Glucocorticoides
4.6. Síndrome de reconstitución inmune
4.7. Ideas claves
El tratamiento de un paciente infectado por VIH con sarcoma de Kaposi (SK) consiste, en primer lugar, en la introducción u optimización del tratamiento antirretroviral (TAR) hasta lograr la supresión virológica. Existen, además tratamientos específicos, locales y sistémicos, cuya indicación va a depender de la extensión de la enfermedad, de la rapidez de crecimiento del tumor, la condición médica del paciente y su estado inmunovirológico.
Tratamiento anirretroviral
Es el tratamiento de elección en todos los pacientes con SK y a veces el único necesario para lograr la remisión en enfermedades no extensas1. Múltiples estudios observacionales han demostrado que desde la introducción del TAR se ha producido una disminución en la incidencia, gravedad y mortalidad del SK en pacientes infectados por VIH2.
Aunque estudios iniciales sugirieron que los inhibidores de la proteasa (IP) podrían tener un efecto antiangiogénico en ratones con SK3, este hecho no se ha constatado en humanos; siendo la supresión virológica, y no el tipo de combinación usada, la que se ha asociado con remisión4. Por tanto, cualquier combinación aprobada para el control de la infección por VIH puede ofrecerse a los pacientes con SK5.
Terapias tópicas
Se utilizan cuando la enfermedad está localizada. Su indicación es principalmente cosmética o ante lesiones locales sintomáticas, pero no previenen la aparición de nuevas lesiones.
4.2.1. Vinblastina tópica: Las inyecciones intralesionales de vinblastina logran un efecto modesto, con aclaramiento de las mismas, aunque sin lograr su resolución completa la mayor parte de las La dosis recomendada, que se puede repetir cada 2 o 3 semanas, es de 0,2-0,3 mg por cada 0,1 ml, debiendo administrar 0,1 ml por cada 0,5 cm 2 de lesión. Como efectos adversos se han descrito neuropatía y alteraciones hematológicas6.
4.2.2. Radioterapia: Se puede usar para tratar lesiones locales extensas o como coadyuvante de la quimioterapia sistémica en lesiones La respuesta suele ser buena, aunque no está exenta de efectos secundarios a nivel local, como irritación o edema7.
4.2.3. Alitretinoina: En ensayos clínicos ha demostrado una respuesta superior al placebo con remisión de las lesiones en un 35-37% vs 7-18% 89 . Su presentación en gel permite la aplicación por el propio
4.2.4. Cirugía: Se ha observado la curación sin recidiva con la cirugía de lesiones localizadas, si bien la evidencia es aún escasa10.
Terapias sistémicas
La administración conjunta de TAR más quimioterapia, comparado con sólo TAR, ha demostrado mayores tasas de remisión del SK en pacientes VIH+10.
Sin embargo, el aumento de toxicidad limita su indicación a pacientes con afectación visceral, enfermedad cutánea extensa, síndrome de reconstitución inmune, edema importante o progresión de la enfermedad a pesar del buen control inmunovirológico11, 12.
4.3.1. Antraciclinas liposomales: Tanto la doxorubicina liposomal (a dosis de 20 mg/m2 cada 3 semanas) como la daunorubicina liposomal (40 mg/m 2 cada 2 semanas), constituyen la quimioterapia de elección para el SK extendido.
Ambas han demostrado en estudios randomizados ser más eficaces y menos tóxicas que otras quimioterapias convencionales (doxorubicina/bleomicina/vincristina)131415. El número mínimo de ciclos a administrar es tres y el máximo depende de la respuesta11. La cardiotoxicidad es el efecto secundario más grave aunque generalmente aparece con dosis acumuladas altas (> 400-450 mg/m 2 ). Para prevenirla se recomienda la realización de un ecocardiograma basal y durante la evolución16.
4.3.2. Paclitaxel: A dosis de 100 mg/m2 cada 2 semanas, ha demostrado tener un efecto similar a las antraciclinas liposomales pero presenta más toxicidad y tiene el inconveniente de metabolizarse por el citocromo P450 y precisar premedicación con glucocorticoides17. Se utiliza sobre todo en países en vías de desarrollo, donde las antraciclinas liposomales no están disponibles, o como segunda línea de
4.3.3. Pomalidomida: Está aprobado por la FDA en pacientes con SK que han fracasado a la combinación de TAR y quimioterapia Esta recomendación se basa en un estudio fase I/II en el que se incluyeron 18 pacientes infectados por VIH, de los cuales 12 (67%) respondieron al tratamiento, siendo una respuesta completa en 3 de ellos18.
Otros fármacos utilizados, con escasa evidencia, han sido el etopósido (demostrando menos efecto que paclitaxel), la vinorelbina o el interferón1112.
Tratamiento antiviral frente al VHH-8
Con el tratamiento del SK no erradicamos el VHH-8, por tanto, el seguimiento estrecho de los pacientes es fundamental, vigilando la aparición de recidivas del SK u otras enfermedades asociadas a este virus, como la enfermedad de Castleman y otros linfomas asociados. Sin embargo, actualmente no existen datos que avalen el uso de fármacos erradicadores del VHH-8 como parte del tratamiento del SK.
Glucocorticoides
En estudios observacionales y de casos y controles, los glucocorticoides se han asociado en pacientes con SK con progresión de la enfermedad y mayor mortalidad, por tanto, su uso se debe evitar siempre que sea posible19.
Síndrome de reconstitución inmune
El síndrome de reconstitución inmune (SRI) consiste en la aparición o empeoramiento de algunas enfermedades oportunistas en pacientes VIH+ tras el inicio del TAR. Se produce por un desequilibrio en la respuesta inmunológica del paciente cuando el TAR empieza a actuar. En pacientes con SK es una complicación frecuente que se ha descrito entre un 6 y un 61% de los casos según las series20. Aparece con más frecuencia en pacientes con estadíos más avanzados de la enfermedad (T1), con cargas virales de VHH-8 más elevadas, en pacientes africanos y al iniciar tratamiento sólo con TAR, sin quimioterapia21.
Cuando aparece se debe añadir al TAR quimioterapia sistémica y evitar los glucocorticoides, ya que, como se ha comentado previamente, se asocian con progresión del SK.
Ideas claves
- Todo paciente con SK debe recibir TAR óptimo. Cualquier combinación incluida en las guías de tratamiento antirretroviral que logre la respuesta virológica es válida (A-II).
En pacientes con afectación visceral, enfermedad cutánea extensa, síndrome de reconstitución inmune, edema importante o progresión de la enfermedad a pesar de tener buen control de la infección por VIH con TAR, está indicado añadir quimioterapia sistémica (A-II).
La quimioterapia de elección para el SK es la doxorubicina o daunorubicina liposomal (A-I).
El paclitaxel es una alternativa a las antraciclinas, cuando éstas no se puedenutilizar o como segunda línea de tratamiento si existe progresión (B-II).
Para enfermedad localizada incapacitante física o estéticamente o como coadyuvante del tratamiento sistémico, se pueden utilizar terapias locales como vinblastina intralesional, radioterapia o cirugía local (A-II).
El uso de glucocorticoides se debe evitar en pacientes con SK pues se asocia con progresión de la enfermedad y aumento de mortalidad (A-II).
Bibliografía:
Martín-Carbonero L, Barrios A, Saballs P, et al. Pegylated liposomal doxorubicin plus highly active antiretroviral therapy versus highly active antiretroviral therapy alone in HIV patients with Kaposi's AIDS 2004; 18: 1737-40.
Grabar S, Abraham B, Mahamat A, et Differential impact of combination antiretroviral therapy in preventing Kaposi´s sarcoma with and without visceral involvement. J Clin Oncol 2006; 24: 3408-14.
Sgadari C, Brillari G, Toschi E, et HIV protease inhibitors are potent anti-angiogenic
Martínez V, Caumes E, Gambotti L, et Remission from Kaposi's sarcoma on HAART is associated with suppression of HIV replication and is independent of protease inhibitor therapy. Br J Cancer 2006; 94 (7):1000-6.
En:http://gesidaseimc.org/wpcontent/uploads/2020/07/TAR_GUIA_GESIDA_2020_ pdf
Saka B, Kombaté K, Mouhari-Toure A, et al. Evaluation of the treatment of Kaposi´s sarcoma with vinblastin in Togo: a study of 23 Bull Soc Pathol Exot; 2011; 104: 339- 41.
Donato V, Guarnaccia R, Dognini J, et Radiaton therapy in the treatment of HIV-
Walmsley S, Northfelt D, Melosky B, et Treatment of AIDS-related cutaneous Kaposi's Sarcoma with topical alitretinoin (9-cis-retinoic acid) gel. Panretin Gel North American Study Group. J Acquir Immune Defic Syndr. 1999 ;22(3): 235-46.
Bodsworth N, Bloch M, Bower M, et Phase III vehicle-controlled, multi-centered study of topical alitretinoin gel 0.1% in cutaneous AIDS-related Kaposi's sarcoma. Am J Clin Dermatol. 2001; 2(2): 77-87.
Gbabe O, Okwundu C, Dedicoat M, et Treatment of severe or progressive Kaposi´s sarcoma in HIV-infected adults. Cochrane Database Syst Rev 2014; 13(8): CD003256.
Reid E, Suneja G, Ambinder R, et al. AIDS-related Kaposi Sarcoma, Version 2.2019, NCCN Clinical Practice Guidelines in J Natl Compr Canc Netw 2019; 17:171-189.
Bower M, Palfreeman A, Alfa-Wali M, et British HIV Association guidelines for HIV- associated malignancies 2014; HIV Med 2014;15:1- 92.
Northfelt DW, Dezube BJ, Thommes JA, et Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi's sarcoma: results of a randomized phase III clinical trial. J Clin Oncol 1998; 16:2445-51.
Stewart S, Jablonowski H, Goebel FD, et Randomized comparative trial of pegylated liposomal doxorubicin versus bleomycin and vincristine in the treatment of AIDS-related Kaposi's sarcoma. International Pegylated Liposomal Doxorubicin Study Group. J Clin Oncol1998; 16: 683-91.
Gill PS, Wernz J, Scadden DT, et Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi's sarcoma. J Clin Oncol 1996; 14: 2353-64.
DOXIL® (doxorubicin hydrochloride liposome injection), for intravenous Horsham, PA: Janssen Products, LP; 2016. Available at: https://www.accessdata.fda.gov/drugsatfda_ docs/label/2016/050718s05 1lbl.pdf.Accessed September 21, 2020.
Cianfrocca M, Lee S, Von Roenn J, et Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma: evidence of symptom palliation from chemotherapy. Cancer 2010; 116(16): 3969- 77.
En:https://accessdata.fda.gov/drugsatfda_docs/label/2020/204026s 023lbl.pdf (Accessed on Oct 1, 2020).
Fernández-Sánchez M, Iglesias MC, Ablanedo-Terrazas Y, et al. Steroids are a risk factor for Kaposi sarcoma-immune reconstitution inflammatory syndrome and mortality in HIV AIDS 2016; 30: 909–914.
Bower M, Nelson M, Young A, et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol 2005; 23:5224-8.
Letang E, Lewis JJ, Bower M; et al. Immune reconstitution inflammatory syndrome associated with Kaposi sarcoma: higher incidence and mortality in Africa than in the UK. AIDS 2013; 27: 1603–1613.
5. LINFOMA PRIMARIO DE CAVIDADES
5.1. Introducción
5.2. Manifestaciones clínicas
5.3. Diagnóstico
5.4. Pronóstico
5.5. Tratamiento
5.6. Ideas claves
Introducción
El linfoma primario de cavidades (LPC) es un linfoma no hodgkiniano (LNH) agresivo de línea B con una presentación clínica y un perfil genético especial. Se describió inicialmente como un linfoma en cavidades pericárdica, pleural o peritoneal en pacientes con infección por el virus de la inmunodeficiencia humana (VIH). Sin embargo, más tarde se constató que puede afectar otras áreas extracavitarias. El virus herpes humano tipo 8 (VHH-8) es el agente etiológico del LPC y el 80% de pacientes están coinfectados por el virus de Epstein-Barr (VEB).
El VHH-8 facilitaría la evasión de las células neoplásicas de los mecanismos normales de inmunovigilancia. Los pacientes con LPC pueden presentar de forma concomitante sarcoma de Kaposi (SK) o enfermedad de Castleman multicéntrica (ECM), dado que comparten una misma causa. Su incidencia puede estar aumentando en la era del tratamiento antirretroviral (TAR) moderno12345.
Manifestaciones clínicas
El LPC suele observarse entre la cuarta y quinta décadas de la vida en pacientes infectados por el VIH, mientras que en individuos no inmunodeprimidos suele observarse entre la séptima y octava décadas. Constituye el 1%-4% de los linfomas asociados a la infección por el VIH. Es más frecuente en hombres que 10 tienen sexo con hombres (HSH). En un tercio de casos existen antecedentes de SK o de ECM. El LPC se caracteriza clínicamente por derrames neoplásicos en las cavidades pleural, pericárdica y peritoneal. Típicamente sólo una cavidad está implicada. Las manifestaciones clínicas más frecuentes son disnea, taponamiento cardiaco o distensión abdominal. Los escasos pacientes con LPC extracavitario afectan al tubo digestivo, piel, pulmones sistema nervioso central y ganglios linfáticos. El recuento de linfocitos CD4+ suele estar muy disminuido en el momento del diagnóstico12345.
Diagnóstico
El estudio citológico de los derrames serosos o de las biopsias de masas tumorales permite establecer el diagnóstico. Aunque en la clasificación de los tumores de la OMS el LPC se definía como un linfoma primario de cavidades, en la reciente actualización se reconoce la variante extracavitaria del PEL. Las células linfomatosas son pleomórficas, con características propias de inmunoblastos, plasmablastos o células anaplásicas. Expresan CD45 y HLA-DR, pero con frecuencia no expresan marcadores B como CD19, CD20, CD79a y PAX5. Expresan marcadores de activación como CD30 y CD38, así como marcadores de diferenciación plasmocelular como CD138, MUM1/IRF4 y BLIMP.
Pueden presentar expresión aberrante de CD3, CD2, CD4, o CD5. Se constata reordenamiento de los genes de inmunoglobulinas. Todo lo anterior sugiere que este linfoma probablemente deriva de un estadio de transición entre células del centro germinal a células post-germinales con diferenciación plasmocelular 67.
Mediante inmunohistoquímica se demuestra infección de las células linfomatosas por VHH-8 con un patrón de latencia, y más raramente de forma lítica, lo que resulta esencial para el diagnóstico. En muchos casos se constata coinfección por el VEB, con un patrón de latencia de tipo 18, 9.
El estudio de extensión es el propio de todo linfoma, e incluye pruebas de imagen con realización de tomografía de emisión de (PET-TC), biopsia de medula ósea y estudio del líquido cefalorraquídeo. Además, es importante estudiar si hay coinfección por virus de la hepatitis B o C.
Pronóstico
En la serie más amplia, que incluye 28 pacientes con LPC la mediana de supervivencia global fue de 6,2 meses con una probabilidad de supervivencia al año del 39%. Los datos sobre factores pronósticos son muy escasos, habida cuenta de que el número de pacientes de cada serie es limitado. El mal estado general, la ausencia de TAR previo a la aparición del linfoma, la afección simultánea de más de una cavidad y la afección pleural o peritoneal respecto a la pericárdica se han identificado como factores pronósticos desfavorables en algunas series. El empleo de TAR potente se ha asociado a un pronóstico más favorable10111213.
Tratamiento
5.5.1 Tratamiento convencional: Las pautas tipo CHOP (Ciclofosfamida, Adriamicina, Vincristina, Prednisona) o las infusionales tipo EPOCH (Etopósido, Prednisona, Vincristina, Ciclofosfamida y Adriamicina) con dosis ajustadas (DA- EPOCH) deparan una tasa de respuesta completa (RC) muy pobre (10%), con una mediana de supervivencia global (SG) de 6 La incorporación de metotrexato a estas pautas ha aumentado ligeramente la frecuencia de RC (30%) y la mediana de SG (10-12 meses) Tabla 1 10111213. La falta de expresión de CD20 en la mayor parte de casos impide el uso de rituximab. Existe escasa información sobre la consolidación con altas dosis de quimioterapia seguida de autotrasplante de progenitores hematopoyéticos, pero los resultados no son prometedores. A su vez, existen casos anecdóticos de trasplante alogénico de progenitores hematopoyéticos, también con resultado escaso.
Como ya se ha comentado, el empleo simultáneo de tratamiento antirretroviral potente ha mejorado los resultados del tratamiento. De hecho, hay casos anecdóticos de respuesta del LPC al instaurar tratamiento antirretroviral o tras la administración de cidofovir, ganciclovir o valganciclovir. Al igual que en otros pacientes sometidos a tratamiento antineoplásico, no debe olvidarse la profilaxis de las infecciones oportunistas, así como el posible empleo de factor estimulante de granulocitos (G-CSF) para acortar el periodo de neutropenia asociada a la quimioterapia.
Por último, los progresos observados en el tratamiento de linfomas con inmunoterapia con anticuerpos monoclonales anti-PD1/PDL1, anti CD30 y anti CD38, 14 entre otros, deberán ser investigados en pacientes con LPC en un futuro próximo.+
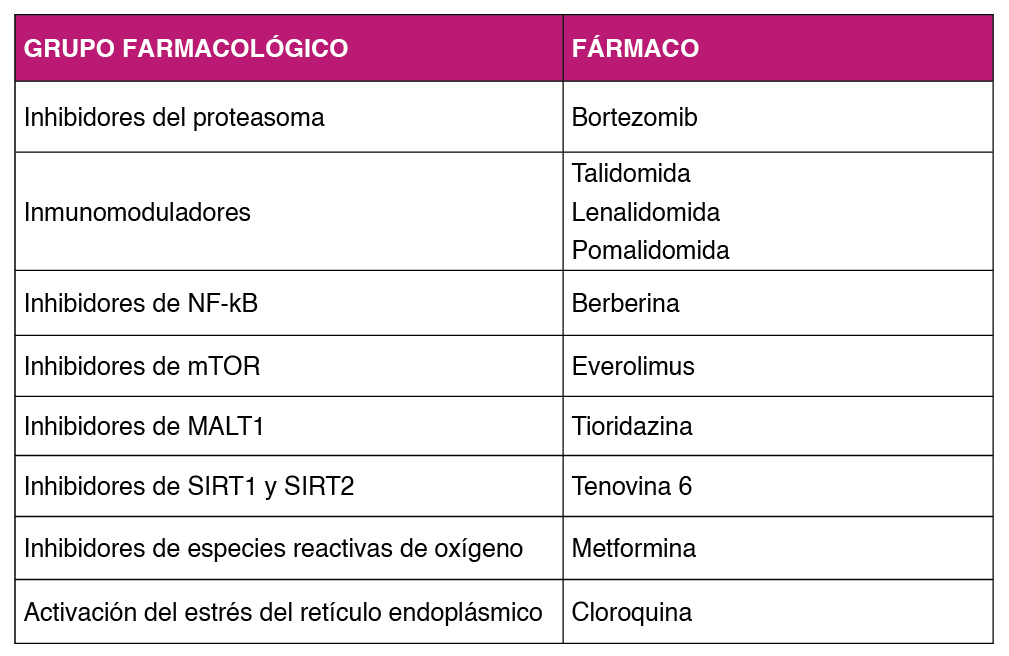
5.5.2 Nuevos tratamientos: Dada la mala respuesta a los tratamientos anteriores, se están evaluando fármacos activos frente a vías de señalización celular como NF-kB, JAK/STAT, and fosfatidilinositol 3-kinasa (PI3K)/AKT Tabla 2. Entre ellos cabe citar el bortezomib, agentes inmunomoduladores (talidomida, lenalidomida y pomalidomida), así como los inhibidores de BET y de m-TOR. La actividad in vitro de bortezomib no parece haberse demostrado in vivo. Respecto a los fármacos inmunomoduladores, se están investigando en ensayos clínicos, en combinación con DA-EPOCH, y de hecho es el estudio en fase de reclutamiento activo más El resto de fármacos están en fases preclínicas de investigación.
Ideas claves
- El linfoma primario de cavidades es un linfoma de línea B poco frecuente en pacientes infectados por el VIH (1%-4% de los linfomas en pacientes VIH positivos). Cursa con derrames serosos y más raramente como masas tumorales ganglionares o extraganglionares.
- La infección por el virus herpes humano 8 es constante y se emplea para el diagnóstico. Puede coexistir infección por el virus de Epstein-Barr.
- Tras el diagnóstico de un LPC asociado al VIH, además del tratamiento quimioterápico se debe iniciar TAR o modificar el previo si existe fracaso (C-II).
La respuesta al tratamiento con pautas convencionales de linfomas agresivos es mala y el pronóstico es desfavorable. La adición de fármacos activos frente a vías de señalización celular está en vías de investigación.
Tablas:

NE: no especificado
Bibliografía:
Arora N, Gupta A, Sadeghi N. Primary effusion lymphoma: current concepts and Curr Opin Pulm Med. 2017; 23:365-370.
Narkhede M, Arora S, Ujjani Primary effusion lymphoma: current perspectives. Onco Targets Ther. 2018; 11:3747-3754.
Shimada K, Hayakawa F, Kiyoi H. Biology and management of primary effusion Blood. 2018; 132:1879-1888.
Lurain K, Polizzotto MN, Aleman K, Bhutani M, Wyvill KM, Gonçalves PH et al. Viral, immunologic, and clinical features of primary effusion Blood. 2019; 133:1753- 1761.
Hübel K. The Changing Landscape of Lymphoma Associated with HIV Infection. Curr Oncol Rep. 2020; 22:111.
Aguilar C, Laberiano C, Beltran B, Diaz C, Taype-Rondan A, Castillo Clinicopathologic characteristics and survival of patients with primary effusion lymphoma. Leuk Lymphoma. 2020; 61:2093-2102.
Said J, Cesarman E. Primary effusion lymphoma. En: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press;
Sukswai N, Lyapichev K, Khoury JD, Medeiros LJ. Diffuse large B-cell lymphoma variants: an update. 2020; 52:53-67.
Verga L, Leni D, Cazzaniga G, Crosta S, Seminati D, Rossi M et The spectrum of the cytopathological features of primary effusion lymphoma and human herpes virus 8-related lymphoproliferative disorders. Cytopathology. 2020; 31:541-546.
Olszewski AJ, Fallah J, Castillo Human immunodeficiency virus-associated lymphomas in the antiretroviral therapy era: analysis of the National Cancer Data Base. Cancer.2016;122:2689-2697.
Guillet S, Gerard L, Meignin V, Agbalika F, Cuccini W, Denis B et al. Classic and extracavitary primary effusion lymphoma in 51 HIV-infected patients from a single Am J Hematol. 2016; 91:233-237.
Boulanger E, Gerard L, Gabarre J, Molina JM, Rapp C, Abino JF et al. Prognostic factors and outcome of human herpesvirus 8-associated primary effusion lymphoma in patients with J Clin Oncol. 2005; 23:4372-4380.
Simonelli C, Spina M, Cinelli R, Talamini R, Tedeschi R, Gloghini A et Clinical features and outcome of primary effusion lymphoma in HIV-infected patients: a single institution study. J Clin Oncol. 2003;21: 3948-3954.
Shah NN, Singavi AK, Harrington Daratumumab in Primary Effusion Lymphoma. N
Bhatt S, Ashlock BM, Toomey NL, Diaz LA, Mesri E, Lossos I et al. Efficacious proteasome/HDAC inhibitor combination therapy for primary effusion lymphoma. J Clin Invest 2013; 123: 2616-28.
Oliveri V, Lanza V, Milardi D, Viale M, Maric I, Sgarlata C et al. Amino- and chloro-8- hydroxyquinolines and their copper complexes as proteasome inhibitors and antiproliferative Metallomics. 2017; 9: 1439-1446.
6. ENFERMEDAD DE CASTLEMAN Y SINDROME INFLAMATORIO INDUCIDO POR CITOKINAS (KICS)
6.1. Introducción
6.2. Epidemiología
6.3. Fisiopatología
6.4. Clínica y laboratorio
6.5. Técnicas de imagen
6.6. Anatomía patológica
6.7. Síndrome inflamatorio inducido por citokinas (KICS)
6.8. Otros pacientes asociados
6.9. Tratamiento
6.10. Ideas claves
Introducción
La enfermedad de Castleman (EC) es un desorden inflamatorio ganglionar que condiciona una hiperplasia folicular angiomatosa. Fue descrita en 1956 por Benjamin Castleman. Existen dos variantes: una variante unicéntrica y otra multicéntrica. La unicéntrica afecta a un único ganglio con patrón de afectación histológica hialinovascular. La multicéntrica afecta a múltiples cadenas ganglionares, pudiendo afectar también a otros órganos, en su anatomía patológica predominan las células plasmáticas (plasmocelular), y se acompaña de manifestaciones inflamatorias sistémicas debidas a la producción de citoquinas, esencialmente la interleukina-6 (IL-6), cursando con reagudizaciones intermitentes. Esta última variante asociada a la infección por virus del herpes humano tipo 8 (VHH-8), es la forma de afectación característica en pacientes con infección por virus de inmunodeficiencia humana (VIH).
Epidemiología
La enfermedad de Castleman multicéntrica (ECM) asociada a VHH-8 y VIH es una entidad poco común cuya frecuencia se ha visto incrementada desde la aparición y uso de tratamiento antirretroviral de gran actividad (TARGA) 1 , si bien ello ha ido también parejo a un descenso en su mortalidad que es actualmente de entre el 10% al 20% a los dos años del diagnóstico 2 . Es más frecuente en varones (entre el 50 al 65%) y aunque puede darse a cualquier edad suele debutar entre la tercera y quinta décadas de vida. Su distribución geográfica es superponible a la de la infección por VHH-8.
Fisiopatología
La ECM en el paciente con infección por VIH es consecuencia de la infección por el VHH-8. A diferencia de otras patologías relacionadas con la coinfección por este virus en el paciente seropositivo, tales como el Sarcoma de Kaposi (SK) o el linfoma primario de cavidades (LPC), en la ECM los episodios de recrudescencia se producen al reactivarse la replicación del VHH-8, pasando así éste de fase latente a lítica 3 . Esto se refleja en el aumento del valor de la PCR de ADN viral que por término medio suele ser de en torno a dos logaritmos. Esto tiene implicaciones en la evolución y tratamiento de estos pacientes. Esta actividad lítica en el plasmablasto es la que desencadena la cascada citoquínica propia de la activación de la vía de IL-6 humana así como de IL-6 del propio virus siendo todo ello responsable de las manifestaciones sistémicas de la enfermedad.
Clínica y laboratorio
La ECM se define por la afectación ganglionar múltiple que, en orden de frecuencia, suele presentarse en: mediastino, pulmón y cuello. Más del 90% de los pacientes asocian fiebre. Pero además es característica la aparición de otras alteraciones tales como organomegalias, anemia, trombocitopenia o trombocitosis, hipoalbuminemia o hipergammaglobulinemia así como edemas, ascitis, astenia, sudoración y caquexia. La mayor parte de estas manifestaciones son debidas a la liberación citoquínica mediada esencialmente por IL-6 y que en las determinaciones de laboratorio se refleja en la elevación de parámetros como la proteína C reactiva, la velocidad de sedimentación globular o las propias determinaciones de interleuquina (IL-6, IL-10, IL-5, IL-8 y IL-12) 4 .
Técnicas de imagen
En cualquier paciente con diagnóstico de ECM o sospecha, es conveniente la realización de una tomografía computarizada (TC) para estudio de extensión de la enfermedad. La tomografía de emisión de positrones (PET-TC) puede aumentar la sensibilidad si bien ocasionalmente la ECM no tiene avidez por 18F-fluorodeoxiglucosa 56 .
En la radiografía de tórax puede verse una linfangitis intersticial, así como agrandamiento ganglionar si bien este estudio es muy limitado para la completa valoración del paciente 7 .
Anatomía patológica
Es la prueba definitoria en el diagnóstico de esta entidad. La morfología ganglionar se encuentra alterada por la presencia incrementada de células B y células plasmáticas policlonales en el manto folicular. Es característica la aparición de neoformaciones vasculares interfoliculares así como el agrandamiento de los folículos implicados. A pesar de esto, la arquitectura ganglionar está preservada.
Además, para el diagnóstico definitivo y para la diferenciación con la variante idiopática de Castleman multicéntrico, será necesario la detección de VHH-8 mediante inmunohistoquímica. Normalmente se determina LANA-1 (antígeno-1 de latencia nuclear), antígeno presente tanto en fase latente como lítica.
Es característico además en pacientes con ECM la sobreexpresión de IgM policlonal y, normalmente, monotípica para cadenas ligeras lambda.
Síndrome inflamatorio inducido por citokinas (KICS)
Las manifestaciones inflamatorias responsables de las alteraciones clínicas y analíticas típicas de los brotes en la ECM se han identificado también en pacientes con infección por VHH-8 en los que no se ha objetivado afectación ganglionar histológica y que, por tanto, no presentan ECM. Esta entidad se conoce como síndrome de liberación de citokinas (KICS) asociado a VHH-8, también denominado virus asociado al sarcoma de Kaposi (HVSK).
Tanto en los pacientes con KICS, como en los pacientes con ECM sintomáticos, la inflamación se debe a la interacción de las citokinas liberadas por el huésped (esencialmente por la activación de la vía de IL-6) con las producidas por el propio VHH-8, al entrar éste en fase lítica (esencialmente la propia IL-6 viral o vIL-6) 8 .
Otros pacientes asociados
En torno al 40% de los pacientes diagnosticados de ECM asocian SK 9 . Si bien no es frecuente, es característica la asociación de la ECM con el síndrome de POEMS que cursa con: polineuropatía, organomegalia, endocrinopatía, mielopatía y alteraciones cutáneas.
Los pacientes con ECM tienen un riesgo elevado de desarrollar linfoma no Hodgkin (entre el 15 al 20% de los diagnosticados en algunas series). De ahí que las terapias con rituximab hayan cambiado el curso y el pronóstico de la ECM. Se han publicado en la literatura algunos casos asilados de pacientes con ECM que han desarrollado Linfoma de Hodgkin 1011 .
Además, la ECM por su correlación con la infección viral por VHH-8 y su disregulación inmunológica puede coexistir con un síndrome hemofagocítico lo que en algunas series puede llegar a ocurrir en el 9% de los casos 12 .
Tratamiento
El tratamiento de la ECM debe instaurarse en pacientes con diagnóstico establecido siempre y cuando asocien manifestaciones de actividad inflamatoria tales como fiebre, elevación de la proteína C reactiva y otros reactantes de fase aguda, alteraciones de laboratorio en el recuento plaquetario u otras de las comentadas.
El fármaco de elección para el tratamiento de la ECM es el rituximab. Se recomienda como tratamiento estándar la administración semanal de 375mg/m2 hasta cuatro dosis. Esto se basa en dos estudios prospectivos publicados en 2007 1314 en los que el uso de este fármaco supuso un aumento significativo de supervivencia tanto por el control de la enfermedad como por la disminución del riesgo de transformación a linfoma, lo que posteriormente ha sido refrendado en sucesivos trabajos 1516 . Este efecto se consigue con independencia de que las células plasmáticas afectadas por la enfermedad expresen el marcador inmunohistoquímico CD20 ya que el rituximab produce depleción de linfocitos B, reservorio del virus VHH-8, y además reduce la expresión citoquínica activada en la ECM 17 . Un riesgo asociado al uso de rituximab en estos pacientes es la posible exacerbación de las lesiones de SK. En esos casos es recomendable la asociación de doxorubicina liposomal que puede además contribuir a la resolución de la propia ECM 18 .
En pacientes con cuadros graves o en aquellos con fracaso a rituximab es posible emplear la combinación antes mencionada, rituximab y doxorubicina; o preferiblemente rituximab y etopósido que en los estudios ha conseguido mejores resultados. Actualmente, se desaconseja el uso de pautas quimioterápicas tipo CHOP (ciclofosfamida, doxorubicina, vincristina y prednisona) salvo que se documente la coexistencia con linfoma.
Una estrategia útil en casos leves, en paciente con SK o para disminuir el riesgo de recidivas es el uso de antivirales tales como cifofovir, foscarnet o los más empleados ganciclovir y valganciovir, para el control de la infección por VHH-8 19 . No están indicadas otras estrategias de terapias de mantenimiento, como el uso periódico de rituximab.
Recientemente han aparecido anticuerpos monoclonales frente a la IL-6 (Siltuximab) o a su receptor (Tocilizumab) 21 que se emplean en enfermedades autoinmunes y que han sido testados para la enfermedad de Castleman idiopática. Actualmente hay estudios en curso en ECM, a la espera de cuyos resultados podrían valorarse como terapia coadyuvante o alternativa a rituximab en pacientes en los cuales no puede emplearse este fármaco.
El tratamiento antirretroviral de alta eficacia si bien no tiene actividad en el tratamiento de ECM, debe siempre incluirse y mantenerse en el esquema terapéutico de estos pacientes.
Ideas claves
- Se debe iniciar tratamiento para la Enfermedad de Castleman Multicéntrica en pacientes con fenómenos inflamatorios acompañantes tales como fiebre, elevación de PCR o trombocitosis (B-III).
El tratamiento de elección es el rituximab (375mg/m2 hasta cuatro dosis de dispensación semanal) (B-II)
Bibliografía:
Powles T, Stebbing J, Bazeos A, Hatzimichael E, Mandalia S, Nelson M, et The role of immune suppression and HHV-8 in the increasing incidence of HIV-associated multicentric Castleman's disease. Ann Oncol. 2009;20(4):775-9.
Bower M, Newsom-Davis T, Naresh K, Merchant S, Lee B, Gazzard B, et al. Clinical Features and Outcome in HIV-Associated Multicentric Castleman's J Clin Oncol. 2011;29(18):2481-6.
Stebbing J, Adams C, Sanitt A, Mletzko S, Nelson M, Gazzard B, et al. Plasma HHV8 DNA predicts relapse in individuals with HIV-associated multicentric Castleman disease. 2011;118(2):271-5.
Oksenhendler E, Carcelain G, Aoki Y, Boulanger E, Maillard A, Clauvel JP, et al. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric castleman disease in HIV-infected Blood. 2000;96(6):2069-73.
Barker R, Kazmi F, Stebbing J, Ngan S, Chinn R, Nelson M, et FDG- PET/CT imaging in the management of HIV-associated multicentric Castleman's disease. Eur J Nucl Med Mol Imaging. 2009;36(4):648-52.
Polizzotto MN, Millo C, Uldrick TS, Aleman K, Whatley M, Wyvill KM, et 18F-fluorodeoxyglucose Positron Emission Tomography in Kaposi Sarcoma Herpesvirus- Associated Multicentric Castleman Disease: Correlation With Activity, Severity, Inflammatory and Virologic Parameters. J Infect Dis. 2015;212(8):1250-60.
Guihot A, Couderc LJ, Agbalika F, Galicier L, Bossi P, Rivaud E, et al. Pulmonary manifestations of multicentric Castleman's disease in HIV infection: a clinical, biological and radiological Eur Respir J. 2005;26(1):118-25.
Uldrick TS, Wang V, O'Mahony D, Aleman K, Wyvill KM, Marshall V, et An interleukin- 6-related systemic inflammatory syndrome in patients co- infected with Kaposi sarcoma- associated herpesvirus and HIV but without Multicentric Castleman disease. Clin Infect Dis. 2010;51(3):350-8.
Oksenhendler E, Boulanger E, Galicier L, Du MQ, Dupin N, Diss TC, et High incidence of Kaposi sarcoma-associated herpesvirus-related non- Hodgkin lymphoma in patients with HIV infection and multicentric Castleman disease. Blood. 2002;99(7):2331-6.
Molinié V, Diebold J, Périé Hodgkin's disease associated with localized or multicentric Castleman's disease. Arch Pathol Lab Med. 1995;119(3):201.
Zarate-Osorno A, Medeiros LJ, Danon AD, Neiman RS. Hodgkin's disease with coexistent Castleman-like histologic features: A report of three cases. . Arch Pathol Lab 1994;118(3):270-4.
Stebbing J, Ngan S, Ibrahim H, Charles P, Nelson M, Kelleher P, et The successful treatment of haemophagocytic syndrome in patients with human immunodeficiency virus- associated multi-centric Castleman's disease. Clin Exp Immunol. 2008;154(3):399-405.
Gerard L, Berezne A, Galicier L, Meignin V, Obadia M, De Castro N, et Prospective study of rituximab in chemotherapy-dependent human immunodeficiency virus associated multicentric Castleman's disease: ANRS 117 CastlemaB Trial. J Clin Oncol. 2007;25(22):3350-6.
Bower M, Powles T, Williams S, Davis TN, Atkins M, Montoto S, et Brief communication: rituximab in HIV-associated multicentric Castleman disease. Ann Intern Med. 2007;147(12):836-9.
Hoffmann C, Schmid H, Muller M, Teutsch C, van Lunzen J, Esser S, et al. Improved outcome with rituximab in patients with HIV-associated multicentric Castleman disease. 2011;118(13):3499-503.
Gerard L, Michot JM, Burcheri S, Fieschi C, Longuet P, Delcey V, et al. Rituximab decreases the risk of lymphoma in patients with HIV-associated multicentric Castleman Blood. 2012;119(10):2228-33.
Bower M, Veraitch O, Szydlo R, Charles P, Kelleher P, Gazzard B, et al.Cytokine changes during rituximab therapy in HIV-associated multicentric Castleman disease. 2009;113(19):4521-4.
Uldrick TS, Polizzotto MN, Aleman K, Wyvill KM, Marshall V, Whitby D, et Rituximab plus liposomal doxorubicin in HIV-infected patients with KSHV- associated multicentric Castleman disease. Blood. 2014;124(24):3544-52.
Uldrick TS, Polizzotto MN, Aleman K, O'Mahony D, Wyvill KM, Wang V, et al. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus- associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood. 2011;117(26):6977-86.
Lurain K, Yarchoan R, Uldrick Treatment of Kaposi Sarcoma Herpesvirus-Associated
Ramaswami R, Lurain K, Peer CJ, Serquina A, Wang V, Widell A, et Tocilizumab in patients with symptomatic Kaposi sarcoma herpesvirus- associated multicentric Castleman disease. Blood. 2020;135(25):2316-9.
7. CONCLUSIONES
- El virus herpes humano tipo 8 (VHH-8) es un virus linfotrópico, que establece un estado de latencia en linfocitos B y en células del endotelio vascular después de una infección primaria, e intermitentemente experimenta periodos de replicación lítica.
- Se transmite fundamentalmente a través de la saliva, siendo necesario que exista un contacto repetido, como en la interacción madre-hijo y en las relaciones sexuales.
- Es más frecuente en hombres que en La prevalencia varia con la localización geográfica, siendo en África y la Amazonia Brasileña superior al 50%. En áreas no endémicas, es más común en hombres que tienen sexo con hombres (HSH).
- La mayor parte de las infecciones son subclínicas y su asociación con enfermedades malignas es significativamente más alta en pacientes infectados por el VIH.
5.Principales procesos clínicos asociados a infección por VHH-8:
- a. Sarcoma de Kaposi (SK): es un angiosarcoma multifocal que se asocia a la infección crónica por el VHH-8 y es más frecuente en pacientes HSH infectados por el El TARGA constituye una pieza clave en su tratamiento. Cuando la enfermedad está localizada se utilizan terapias tópicas; en caso de enfermedad sistémica, la administración conjunta de TAR más quimioterapia (doxorubicina o daunorubicina), ha demostrado mayores tasas de remisión del SK en pacientes VIH positivos. El uso de glucocorticoides se debe evitar en pacientes con SK pues se asocia con progresión de la enfermedad y aumento de mortalidad. El síndrome de reconstitución inmune en pacientes con SK es una complicación frecuente que se ha descrito entre un 6 y un 61% de los casos.
- b. Linfoma primario de cavidades (LPC): Constituye el 1-4% de los linfomas asociados a la infección por el VIH. Se caracteriza clínicamente por derrames neoplásicos en las cavidades pleural, pericárdica y peritoneal, aunque puede afectar a otras áreas En muchos casos se constata coinfección por el virus de Epstein Barr (VEB). La respuesta al tratamiento con pautas convencionales de linfomas agresivos es mala y el pronóstico es desfavorable. La adición de fármacos activos frente a vías de señalización celular está en vías de investigación.
- c. La enfermedad de Castleman multicéntrica (ECM) es un desorden inflamatorio ganglionar que condiciona una hiperplasia folicular angiomatosa en mediastino, pulmón y cuello, presentando fiebre el 90% de los Es más frecuente en varones VIH entre la tercera y quinta década de vida. El diagnóstico se realiza por pruebas de imagen, anatomía patológica y la detección de VHH-8 por técnicas de PCR. El tratamiento de elección es el rituximab.
- d. Síndrome inflamatorio inducido por citoquinas asociado a VHH-8: Manifestaciones inflamatorias típicas de los brotes de ECM en pacientes con infección por VHH-8 en los que no se ha objetivado afectación ganglionar histológica. La inflamación se debe a la interacción de las citokinas liberadas por el huésped (por la activación de la vía IL-6) con las producidas por el propio VHH-8 al entrar en fase lítica (IL-6 viral o vIL-6). No existen guías específicas para su tratamiento, pero los fármacos empleados son los mismos que los utilizados en la ECM.
8. DECLARACIÓN DE TRANSPARENCIA (CONFLICTOS DE INTERÉS)
- La Dra. Teresa Aldámiz Echevarría-Lois declara haber participado en reuniones científicas y recibidos honorarios de ViiV, MSD y Gilead. En relación con el presente documento no existe ningún conflicto de interés.
La Dra. Cristina Díez Romero declara no presentar ningún conflicto de interés en relación con el presente documento.
La Dra. Carmen Hidalgo declara no presentar ningún conflicto de interés en relación con el presente documento.
- El Dr. Arkaitz Imaz ha disfrutado de ayudas para investigación clínica de Gilead Ssciences, Merck Sharp & Dohme y ViiV Healthcare; ha recibido compensación económica por labores de consultoría para Gilead Sciences y ViiV Healthcare; compensción económica por charlas de Abbvie, Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare; y ha recibido ayudas para asistencia a congresos de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare. No presenta ningún conflicto de interés en relación con el presente documento.
La Dra. Mª Ángeles Marcos declara no presentar ningún conflicto de interés en relación con el presente documento.
La Dra. Luz Martín Carbonero ha recibido pagos por labor de asesoría de los laboratorios VIIV, Jansen, MSD y ha recibido pagos por charlas de los laboratorios VIIV, Gilead, MSD. No presenta ningún conflicto de interés en relación con el presente documento.
La Dra. Pilar Miralles no presenta ningún conflicto de interés en relación con el presente documento.
La Dra. Victoria Moreno Celda no presenta ningún conflicto de interés en relación con el presente documento.
El Dr. Daniel Podzamcer no presenta ningún conflicto de interés en relación con el presente documento.
El Dr. Josep Mª Ribera Santasusana declara no presentar ningún conflicto de interés en relación con el presente documento.
El Dr. José Sanz Moreno no presenta ningún conflicto de interés en relación con el presente documento.
La Dra. Eulalia Valencia Ortega declara haber recibido compensación económica por charlas y labores de asesoría, así como ayuda para asistencia de Congresos por parte de MSD, ViiV Healthcare, Janssen Cilag y Gilead Sciences. No presenta ningún conflicto de interés en relación con el presente documento.
La Dra. Isabel Viciana Ramos declara haber participado en reuniones científicas y recibidos honorarios de ViiV, MSD y Gilead. En relación con el presente documento no presenta ningún conflicto de interés.