Documento de consenso de GeSIDA - Panel de expertos de GeSIDA respecto al tratamiento antirretroviral en adultos infectados por el virus de la inmunodeficiencia humana.
Actualización enero 2025
Notas de la Versión:
COMITÉ DE REDACCIÓN
Coordinadores:
Rosario Palacios |
Hospital Universitario Virgen de la Victoria, IBIMA, Málaga |
María Velasco Arribas |
Hospital Universitario Fundación Alcorcón. Madrid |
Redactores Generales:
Juan González-García |
Hospital Universitario La Paz. IdiPAZ, Madrid |
Rosario Palacios |
Hospital Universitario Virgen de la Victoria, IBIMA, Málaga |
Redactor Novel:
Azucena Bautista Hernández |
Hospital Universitario de la Princesa. IIS La Princesa. Madrid. CIBERINFEC ISCIII. |
Redactores y Revisores:
Teresa Aldamiz-Echevarría Lois |
Hospital General Universitario Gregorio Marañón. IiSGM. Madrid |
José Ramón Arribas |
Hospital Universitario La Paz. IdiPAZ, Madrid |
Alberto Díaz de Santiago |
Hospital Universitario Puerta de Hierro/IDIPHIM. Madrid |
Nuria Espinosa Aguilera |
Hospital Universitario Virgen del Rocío/IBiS. Sevilla |
Juan Flores Cid |
Hospital Arnau de Vilanova. Valencia |
María José Galindo |
Hospital Clínico Universitario. Universidad de Valencia. Valencia |
Federico García |
Hospital Clínico Universitario San Cecilio. Granada |
Arkaitz Imaz |
Hospital Universitari de Bellvitge. IDIBELL. L’Hospitalet de Llobregat. Barcelona |
Josep M.ª Llibre Codina |
Hospital Universitari Germans Trias i Pujol. Badalona/IGTP. Universidad de Barcelona. Barcelona |
Luz Martín-Carbonero |
Hospital Universitario La Paz. IdiPAZ. Madrid |
Esteban Martínez |
Hospital Clinic Universitari. IDIBAPS. Universidad de Barcelona. Barcelona |
José Moltó |
Fundació Lluita contra les Infeccions. Hospital Germans Trias i Pujol de Badalona/IGTP. Barcelona |
Marta Montero |
Hospital Universitario La Fe. Valencia |
Eugenia Negredo |
Fundació Lluita contra les Infeccions. Hospital Germans Trias i Pujol de Badalona/IGTP. Barcelona. CIBERINFEC-ISCIII |
David Rial Crestelo |
Hospital Universitario 12 de Octubre /Instituto de Investigación 1+12. Madrid |
Ezequiel Ruíz-Mateos Carmona |
Laboratorio de Inmunovirología Instituto de Biomedicina de Sevilla (IBiS)/Servicio de Enfermedades Infecciosas. Hospital Virgen del Rocío. Sevilla |
Matilde Sánchez-Conde |
Hospital Ramón y Cajal.Madrid. IRYCIS. CIBERINFEC ISCIII |
Ignacio de los Santos Gil |
Hospital Universitario de la Princesa. IIS La Princesa. Madrid. CIBERINFEC ISCIII. |
José Sanz-Moreno |
Hospital Universitario Príncipe de Asturias. Alcalá de Henares. Madrid |
AGRADECIMIENTOS
La Junta Directiva de GeSIDA agradece las aportaciones y opiniones para mejorar el texto de: Adriá Curran, Jose M. Cisneros, Jose A. Iribarren, Jara Llenas, Juan Emilio Losa, Mª Jesús Pérez Elías, Jesús Santos, Departamento Médico de Gilead Sciences (enviado por Cristina de Alvaro) y Departamento Médico de Merck Sharp and Dohme (enviado por Fernando Chacón, Immaculada Clotet, Manuel Cotarelo, Laura Martín y Enrique Vacas).
ABREVIATURAS UTILIZADAS
/ |
Signo entre dos principios activos indica coformulación |
+ |
Signo entre dos principios activos indica formulaciones separadas |
3TC |
Lamivudina |
ABC |
Abacavir |
AP |
Acción Prolongada |
ARV |
Antirretrovirales |
ATV |
Atazanavir |
BID |
Fármaco o pauta terapéutica administrados dos veces al día |
BIC |
Bictegravir |
/c |
Potenciado con cobicistat |
CAB |
Cabotegravir |
CAP |
Parámetro de atenuación controlada |
CAPD |
Diálisis peritoneal |
CMV |
Citomegalovirus |
COBI |
Cobicistat |
Cop/mL |
Copias/mililitro |
CVP |
Carga viral plasmática |
CYP3A |
Citocromo P450 3A |
DMO |
Densidad mineral ósea |
DOR |
Doravirina |
DRV |
Darunavir |
DTG |
Dolutegravir |
FGe |
Filtrado glomerular estimado |
EA |
Efecto adverso |
EC |
Ensayo Clínico |
EFV |
Efavirenz |
eHMET |
Enfermedad Hepática Metabólica |
EMA |
European Medicines Agency |
ENF |
Enfuvirtide, T20 |
ETR |
Etravirina |
EVG |
Elvitegravir |
FAR |
Fármacos antirretrovirales |
FRAX |
Riesgo de fractura por fragilidad ósea |
FTC |
Emtricitabina |
FTV |
Fostemsavir |
FV |
Fracaso virológico |
GeSIDA |
Grupo de estudio de SIDA |
G6PD |
Glucosa 6 fosfatodeshidrogenasa |
HD |
Hemodiálisis |
IBA |
Ibalizumab |
HSH |
Hombre con relaciones sexuales con hombres |
INI |
Inhibidor de la integrasa |
IM |
Intramuscular |
IO |
Infección oportunista |
IP |
Inhibidor de la proteasa |
IP/p |
Inhibidor de la proteasa potenciado |
ITIAN |
Inhibidor transcriptasa inversa análogo de nucleósido/nucleótido. |
ITINN |
Inhibidor de transcriptasa inversa no nucleósido |
ITS |
Infecciones de transmisión sexual |
ITT |
Análisis por intención de tratar |
LEN |
Lenacapavir |
LPV |
Lopinavir |
Mx |
Moxifloxacino |
MR |
Mutaciones de resistencia |
MVC |
Maraviroc |
NGS |
Secuenciación de Nueva Generación |
NPJ |
Neumonía por Pneumocystis jirovecii |
NVP |
Nevirapina |
OMS |
Organización Mundial de la Salud |
PPD |
Prueba de la tuberculina, Mantoux |
PNS |
Plan Nacional sobre el SIDA |
PrEP |
Profilaxis pre-exposición |
PVV |
Personas que viven con VIH |
QD |
Fármaco o pauta de tratamiento administrada una vez al día |
RAL |
Raltegravir |
RCV |
Riesgo cardiovascular |
RHS |
Reacción de hipersensibilidad |
RP |
Rifapentina |
RPV |
Rilpivirina |
/r |
Potenciado con ritonavir |
RTV |
Ritonavir |
SEIMC |
Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica |
SIDA |
Síndrome de inmunodeficiencia adquirida |
SIRI |
Síndrome inflamatorio de reconstitución inmune |
SNC |
Sistema nervioso central |
TAF |
Tenofovir alafenamida |
TAMs |
Mutaciones asociadas con resistencia a los análogos de la timidina |
TAR |
Tratamiento antirretroviral; ídem. de alta eficacia |
TB |
Tuberculosis |
TDF |
Tenofovir disoproxil fumarato |
TDx |
Tenofovir disoproxil |
TFV |
Tenofovir (en cualquiera de sus presentaciones) |
TLOVR |
Tiempo hasta la pérdida de la respuesta virológica (time to loss of virologic response) |
TO |
Tratamiento optimizado |
UGT |
UDP-Glucuroniltransferasa |
VBN |
Viremia bajo nivel |
VIH-1 |
Virus de la inmunodeficiencia humana tipo 1 |
VIH-2 |
Virus de la inmunodeficiencia humana tipo 2 |
VH (A,B,C,D) |
Virus de la hepatitis (A, B, C, Delta) |
XTC |
3TC o FTC indiferentemente |
ZDV |
Zidovudina |
DECLARACIÓN DE TRANSPARENCIA (CONFLICTOS DE INTERÉS )
██ Teresa Aldamiz-Echevarria ha recibido honorarios por participar en reuniones científicas de Gilead, MSD, ViiV y Janssen.
██ Jose Ramón Arribas ha efectuado labores de consultoría para los laboratorios Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme, ViiV Healthcare, Teva, Alexa y Serono; ha disfrutado de becas para investigación clínica de ViiV Healthcare y Gilead Sciences; y ha recibido compensación económica por ponencias de Merck Sharp & Dohme y ViiV Healthcare.
██ Azucena Bautista Hernández ha recibido compensación económica por ponencias de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare; ha recibido ayudas para la asistencia a congresos y cursos formativos de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
██ Ignacio de los Santos ha recibido pagos por ponencias de Janssen, VIIV, Gilead y MSD.
██ Alberto Díaz de Santiago ha recibido financiación para investigación de Gilead Sciences, ViiV Healthare y Janssen pharmaceuticals. Ha participado en Advisory Boards para Gilead Sciences, ViiV Healthcare, MSD y Janssen pharmaceuticals. Ha recibido compensación económica por ponencias de Gilead Sciences, ViiV Healthcare, MSD y Janssen pharmaceuticals.
██ Nuria Espinosa, He participado en advisories y colaborado en eventos de Gilead ,Viivh y Janssen. He recibido financiación para proyectos de investigación de Merck.
██ Juan Flores ha recibido honorarios por ponencias de los laboratorios Gilead, VIIV, MSD y Janssen.
██ Mª. José Galindo ha recibido honorarios de ViiV Healthcare, Johnson and Johnson, MSD y Gilead por realizar presentaciones, materiales educativos y asesorías.
██ Federico García ha recibido ayudas para proyectos de investigación de Gilead , ViiV, MSD y Roche; honorarios por ponencias de ViiV, Gilead, Abbvie, MSD, Janssen, Roche, Biomerieux, Hologic, Qiagen; ha recibido ayudas para asistencia a actividades formativas de ViiV, Gilead, Abbvie, MSD, Janssen, Roche, Biomerieux, Hologic, Qiagen; Labores de asesoramiento para ViiV, Gilead, Abbvie, Roche y Hologic.
██ Juan González-García ha efectuado labores de consultoría para ViiV. Ha recibido ayudas para investigación y compensaciones económicas por ponencias de Gilead Sciences, Merck Sharp & Dohme y ViiV Healthcare. Ha recibido igualmente ayudas económicas para asistir a reuniones científicas de Gilead Sciences.
██ Arkaitz Imaz ha recibido compensación económica por ponencias, actividades formativas y/o participación en consultorías, así como becas para investigación y ayudas para asistencia a congresos científicos de Gilead Sciences, Johnson&Johnson, Merck Sharp & Dohme TheraTechnologies y ViiV Healthcare;
██ Josep M Llibre ha realizado labores de asesoría o formación continuada con ViiV Healthcare, Gilead Sciences y MSD y ha sido testigo experto para Gilead Sciences.
██ Luz Martín Carbonero ha efectuado labores de consultoría para ViiV, Gilead Sciences. Ha recibido compensaciones económicas por ponencias de Gilead Sciences, Janssen y ViiV Healthcare. Ha recibido igualmente ayudas económicas para asistir a reuniones científicas de Gilead Sciences.
██ Esteban Martínez ha recibido honorarios por realización de actividades educativas médicas o participación en consejos asesores de Gilead Sciences, Janssen Cilag, MSD y ViiV Healthcare. Su institución ha recibido becas de Gilead Sciences, MSD y ViiV Healthcare para proyectos de investigación clínica que lidera.
██ José Moltó he recibido financiación para proyectos de investigación, honorarios por consultorías y patrocinios de conferencias de varios laboratorios (MSD, Gilead Sciences, Viiv Healthcare, Janssen Cilag).
██ Marta Montero ha recibido honorarios y/o ayudas para actividades con fines docentes o ponencias de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme, Menarini y ViiV Healthcare.
██ Eugènia Negredo ha recibido financiación para investigación, honorarios de consultoría y patrocinio para conferencias de Gilead Science, ViiV Healthcare, Janssen Cilag y MSD, y he formado parte de comités asesores para estas empresas.
██ Rosario Palacios ha efectuado labores de consultoría para los laboratorios Gilead Sciences, Merck Sharp & Dohme y ViiV Healthcare; ha recibido compensación económica por ponencias de Gilead Sciences, Johnson and Johnson, Merck Sharp & Dohme y ViiV Healthcare.
██ David Rial Crestelo ha recibido honorarios por participar en presentaciones y conferencias de Gilead Science y ViiV Healthcare.
██ Ezequiel Ruiz-Mateos Carmona ha recibido proyectos financiados por Gilead Science Inc
██ Matilde Sánchez Conde ha recibido financiación para el desarrollo de proyectos de investigación de Gilead, ViiV y Merck, así como financiación de Gilead para la asistencia a congresos científicos y por impartir ponencias en jornadas científicas.
██ José Sanz Moreno ha recibido compensación económica por labores docentes y ayuda para asistencia a congresos y reuniones científicas de Johnson & Johnson, Merck Sharp & Dohme, ViiV Healthcare y Gilead Sciences.
██ María Velasco ha recibido honorarios y/o ayudas para actividades con fines docentes, ponencias o investigación de Gilead Sciences, Janssen Cilag, Merck Sharp & Dohme y ViiV Healthcare.
1. INTRODUCCIÓN
JUSTIFICACIÓN, OBJETIVO Y ALCANCE
El uso de los fármacos antirretrovirales (FAR) ha adquirido gran complejidad por la disponibilidad de seis familias, incluyendo más de 40 fármacos y combinaciones, y por sus diferentes características en cuanto a eficacia, toxicidad, resistencias, barrera genética, tropismo, interacciones y uso en situaciones clínicas especiales. Esta complejidad hace necesaria la elaboración de guías y recomendaciones sobre el tratamiento antirretroviral (TAR).
El Grupo de Estudio de SIDA (GeSIDA) de la Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC) edita periódicamente un documento de consenso sobre el TAR en adultos.
El objetivo de este documento es transmitir el estado actual del conocimiento sobre el TAR a los profesionales que tratan a adultos con infección por el VIH y proporcionarles recomendaciones que puedan guiar sus decisiones terapéuticas.
GeSIDA, junto con otras sociedades científicas, elabora otras recomendaciones referentes a la infección por el VIH donde se incluyen aspectos específicos del TAR. En este documento estos aspectos se tratan de forma somera y se remite al lector a las publicaciones específicas.
METODOLOGÍA
El panel redactor del documento está integrado por clínicos expertos en la infección por el VIH y el TAR, distribuidos por grupos encargados de actualizar cada sección del documento. Dos miembros del panel actúan como coordinadores y dos como redactores generales, con apoyo, de un redactor novel. Cada grupo revisa los datos más relevantes de las publicaciones científicas y comunicaciones a congresos más recientes, en el caso de esta actualización hasta el 30 de noviembre de 2024, elaboran el texto de cada sección y generan preguntas sobre aspectos no suficientemente consensuados que se someten a votación de todo el panel. El borrador del documento se discute y consensua en una reunión presencial del panel y su redacción provisional se expone durante 15 días en la página web de GeSIDA para que profesionales, pacientes o quien esté interesado pueda hacer sugerencias que, si procede, son integradas en el documento final.
Cada recomendación de estas guías se califica con una letra que indica su fuerza [A (debe ofrecerse siempre), B (en general debe ofrecerse) o C (debe ofrecerse opcionalmente)] y un número que expresa las pruebas que sustentan dicha recomendación [I (resultados de uno o más ensayos clínicos aleatorizados de aspectos clínicos o de laboratorio o de un metaanálisis), II (de uno o más ensayos no aleatorizados o datos observacionales de cohortes) y III (opinión de expertos)].
2. EVALUACIÓN PARA GUIAR EL TRATAMIENTO ANTIRRETROVIRAL
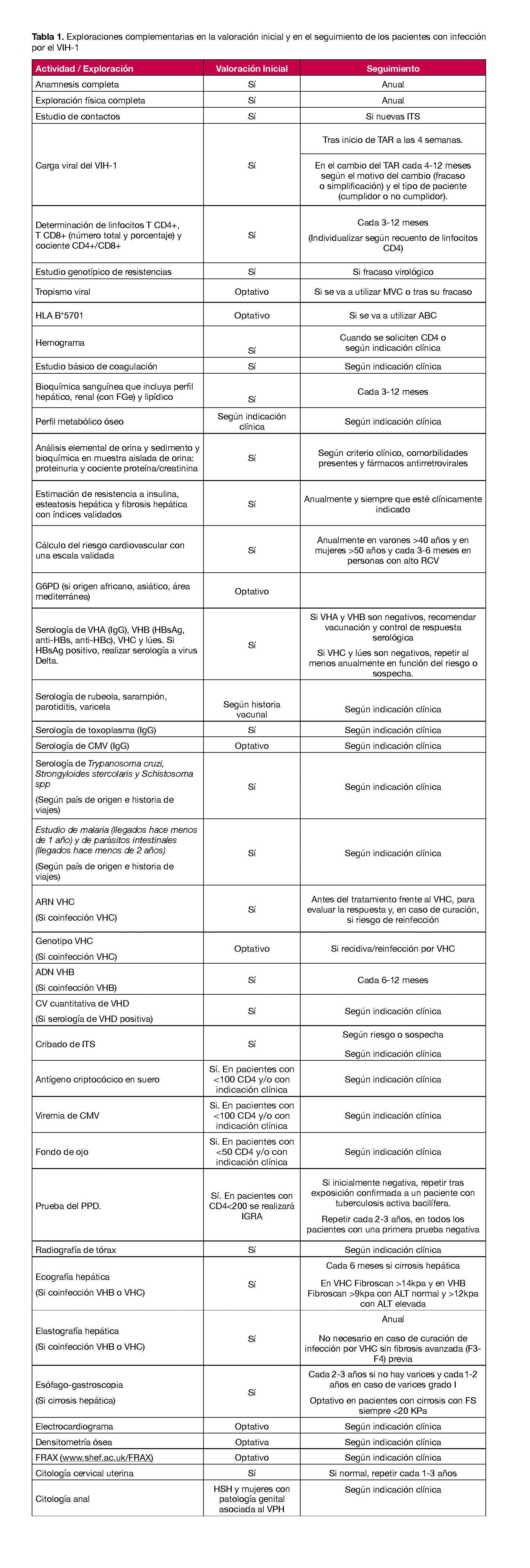
En la Tabla 1 se sistematizan las exploraciones complementarias en la valoración inicial y en el seguimiento de los pacientes con infección por el VIH-1
Recomendaciones
- Realizar una anamnesis detallada y un examen físico completo que se repetirán siempre que el paciente lo requiera y como mínimo anualmente (A-III).1
- Realizar un estudio voluntario de contactos en todos los nuevos diagnósticos de VIH-1, garantizando la confidencialidad (A-III).
- En la visita inicial, realizar una serología del VIH-1/2 en los casos en los que no se haya confirmado la infección o la CVP sea indetectable (A-I).
- Determinar la CVP antes del inicio del TAR, cuando se cambie de TAR y periódicamente durante el tratamiento, con una técnica con un límite de detección de al menos 50 copias/mL, para confirmar y monitorizar la supresión virológica (A-I).
- Determinar la cifra absoluta y el porcentaje de linfocitos CD4+ antes de iniciar el TAR y, una vez iniciado, como parámetro de monitorización periódica de la respuesta inmunológica al mismo (A-I).
- Determinar la cifra de linfocitos CD8+ o el cociente CD4/CD8 cuando se determine la cifra de linfocitos CD4+ en sangre (B-II).
- Se recomienda la realización de un estudio genotípico de resistencias del VIH-1 en todos los pacientes antes del inicio del TAR, lo que no impide el inicio del mismo, aunque no se disponga de dicho estudio en ese momento. (B-II)2
- Esperar a conocer el resultado del estudio genotípico de resistencias si se va a iniciar TAR con una pauta basada en ITNN para los que existe mayor prevalencia de resistencias transmitidas (EFV y RPV) (A-II) y cuando se vaya a iniciar con DTG-3TC en un paciente con historia de uso de PrEP3(A-III). En los demás casos, revisar la prueba en cuanto esté disponible, especialmente si se ha utilizado una pauta de baja barrera genética (A-III).6
- Realizar un estudio genotípico de resistencias del VIH-1 a todos los pacientes en fracaso virológico (A-I).
- Determinar el tropismo del VIH-1 cuando se vaya a utilizar un antagonista del receptor de CCR5 y/o cuando fracase un régimen con un antagonista del receptor CCR5 (A-I).
- Determinar el HLA-B*5701 en todos los pacientes antes de iniciar un régimen de TAR que contenga ABC, evitando la prescripción del mismo si el HLA-B*5701 es positivo (A-I).
- Incluir en la evaluación inicial de laboratorio: hemograma, bioquímica general, estudio básico de coagulación, serologías y pruebas específicas (A-II).
Tablas:
Bibliografía:
Grupo de Estudio de Sida de la Sociedad Española de Enfermedades Infecciosas. Documento consenso de GeSIDA sobre control y monitorización de la infección por el VIH. (Actualización abril 2018). https://gesida-seimc.org/wp-content/uploads/2018/02/gesida_DC_Control_Monitorizacion_VIH.pdf
Viñuela-González L, Fuentes-López A, Serrano-Conde E, et al. Resistencias transmitidas en pacientes naïve. Actualización 2019-2021. XIII Congreso Nacional GeSIDA. 27-30 de noviembre de 2022. Sitges. Abstract PO-24
Viñuela L, de Salazar A, Fuentes A, Serrano-Conde E, Falces-Romero I, Pinto A, Portilla I, Masiá M, Peraire J, Gómez-Sirvent JL, Sanchiz M, Iborra A, Baza B, Aguilera A, Olalla J, Espinosa N, Iribarren JA, Martínez-Velasco M, Imaz A, Montero M, Rivero M, Suarez-García I, Maciá MD, Galán JC, Perez-Elias MJ, García-Fraile LJ, Moreno C, Garcia F. Transmitted drug resistance to antiretroviral drugs in Spain during the period 2019-2021. J Med Virol. 2023 Dec;95(12):e29287. doi: 10.1002/jmv.29287. PMID: 38084763Principio del formulario
De Salazar A, Viñuela L, Fuentes A, Teyssou E, Charpentier C, Lambert-Niclot S, Serrano-Conde E, Pingarilho M, Fabeni L, De Monte A, Stefic K, Perno CF, Aguilera A, Falces I, Delgado R, Fernandes S, Diogo I, Gomes P, Paraskevis D, Santoro MM, Ceccherini-Silberstein F, Marcelin AG, Garcia F. Transmitted Drug Resistance to Integrase-Based First-Line Human Immunodeficiency Virus Antiretroviral Regimens in Mediterranean Europe. Clin Infect Dis. 2023 May 3;76(9):1628-1635. doi: 10.1093/cid/ciac972. PMID: 36571282.
Girometti N, McCormackS, Tittle V, McOwanA, Whitlock G. Rising rates of recent preexposure prophylaxis exposure among men having sex with men newly diagnosed with HIV: antiviral resistance patterns and treatment outcomes. AIDS 2022; 36: 561-566.
Grupo de Estudio de Sida de la Sociedad Española de Enfermedades Infecciosas. Documento sobre la utilidad clínica de las resistencias a antirretrovirales. (Actualización octubre 2023). https://gesida-seimc.org/wp-content/uploads/2024/01/documento_sobre_interpretacion_y_utilidad_clinica.pdf
3. TRATAMIENTO ANTIRRETROVIRAL INICIAL
Los objetivos del TAR son conseguir la máxima y más duradera supresión de la CVP, restablecer y preservar la función inmunológica, reducir la morbilidad asociada a la replicación del VIH-1 y su efecto sobre otras comorbilidades, aumentar la supervivencia y prevenir la transmisión del VIH-1.
GENERALIDADES
3.1.1. CUANDO INICIAR EL TAR
El TAR debe iniciarse tan pronto como sea posible en todas las personas que viven con VIH (PVV), con o sin sintomatología, y con independencia del número de linfocitos T CD4+ Tabla 1. Como excepción se pueden considerar las PVV que mantienen CVP indetectable de forma mantenida sin TAR (controladores de élite). La existencia de ciertas infecciones concomitantes puede condicionar la inmediatez del comienzo del TAR (véase el apartado "Paciente con evento oportunista"). La situación clínica del paciente, así como su disposición y motivación son factores críticos a la hora de decidir el momento para iniciar el TAR. La recomendación de inicio en todos los pacientes se sustenta sobre todo en dos grandes ensayos clínicos aleatorizados12.
En el ensayo START1, se incluyeron 4.685 PVV infectadas por el VIH-1 con una cifra de linfocitos CD4+ >500 células/μL, y seguidas durante un tiempo medio de 3 años. Se aleatorizaron a iniciar TAR de forma inmediata o diferido hasta un recuento de linfocitos CD4+ <350 células/μL. La variable principal (evento definitorio de SIDA, complicación grave no asociada a SIDA o muerte por cualquier motivo) ocurrió en el 1,8% de los pacientes que iniciaron TAR de forma inmediata y en el 4,1% de los que lo difirieron (reducción de riesgo del 57% [IC95%: 38% - 70%]).
En el estudio TEMPRANO2 se incluyeron 2.056 pacientes sin TAR previo y una cifra de linfocitos CD4+ <800 células/μL que se asignaron aleatoriamente a recibir TAR de forma inmediata o diferido hasta presentar criterios de tratamiento según las recomendaciones de la Organización Mundial de la Salud (OMS) vigentes en cada momento. Con el inicio inmediato del TAR, a lo largo de 30 meses la variable principal (desarrollo de SIDA, cáncer no asociado a SIDA, enfermedad bacteriana invasiva, o muerte por cualquier causa) se redujo en la totalidad de los participantes del estudio (44%; IC95%: 24 - 59%) y en los que entraron con una cifra de linfocitos CD4+ superior a 500 células/μL (44%; IC95%: 6 - 67%).
Por otra parte, se ha demostrado que el inicio del TAR se asocia con un menor riesgo de transmisión del VIH-1 y reducción de nuevas infecciones3.
Aunque varios ensayos clínicos realizados en países con recursos económicos limitados4 han mostrado que el inicio rápido del TAR (el mismo día del diagnóstico o en la primera semana) favorece la retención de los pacientes en la asistencia e incrementa la proporción de pacientes con supresión virológica, en países desarrollados con sistemas sanitarios públicos como el español no existe evidencia de que el inicio inmediato tenga un impacto positivo sobre la retención en los cuidados.
En el estudio DIAMOND, no comparativo, se evaluó en 109 pacientes la eficacia de DRV/c/FTC/TAF iniciado en los primeros 14 días tras el diagnóstico, antes de disponer de las determinaciones basales de laboratorio. A las 48 semanas, el 84% de los pacientes tenían CVP <50 cop/mL en el análisis snapshot, y 96% en el análisis de datos observados (n=96), sin discontinuaciones por fracaso5.
En el estudio STAT, se incluyeron 131 pacientes sin tratamiento previo en un estudio de brazo único con la combinación DTG/3TC, en los primeros 14 días tras el diagnóstico de infección por VIH-1, sin resultados analíticos previos. A lo largo de las primeras 24 semanas, el tratamiento se modificó en 8 pacientes (5 con infección por virus de hepatitis B [VHB], uno con la mutación M184V al inicio, uno por evento adverso y uno por decisión del paciente). A las 24 semanas, el 78% estaban con CVP <50 cop/mL en el análisis snapshot, y a las 48 semanas el 82%6. En los escasos FV, no se detectó emergencia de mutaciones asociadas a resistencias (MR).
En el estudio FAST7 se evaluó la combinación bictegravir (BIC)/FTC/TAF en una estrategia de inicio rápido. Se incluyeron 117 participantes. En el análisis snapshot, a las 24 semanas, el 80,4% estaban con CVP <50 cop/mL, 9,8% con CVP >50 cop/mL, 1,8% habían abandonado por evento adverso o muerte y 8% por otras causas. No se detectaron MR en los pacientes con CVP detectable a las 24 semanas.
En el estudio BIC-NOW, en fase IV, prospectivo, de rama única, multicéntrico y realizado en España, se analizó la respuesta al régimen de BIC/FTC/TAF de inicio inmediato en la primera visita médica especializada, en 208 PVV (22% con CD4+ <200 células/μL, 43,3% con CVP >100.000 cop/mL, 22,6% con criterios de SIDA y 4,3% coinfectados con el VHB). A las 48 semanas el 84,1% y el 98,3% presentaron CVP <50 cop/mL en el análisis snapshot y por protocolo, respectivamente. Se detectó CVP >50 cop/mL en el 1,7% de los participantes (ninguno desarrolló MR), atribuido a mala adherencia, y hubo pérdida de seguimiento en el 9,6%8.
Una revisión sistemática de 4 estudios prospectivos en fase IV ha confirmado la eficacia de BIC/FTC/TAF en el inicio rápido del TAR9.
En un ensayo clínico aleatorizado y abierto, recientemente realizado en China, se comparó el TAR iniciado en los 14 días tras el diagnóstico en 300 participantes, que recibieron EFV (400 mg) +3TC+TDF frente a BIC/FTC/TAF, demostrando con este último régimen eficacia virológica estadísticamente superior a las 48 semanas (79,2% y 95,95%, respectivamente), con más frecuentes discontinuaciones (16,1% y 0,7%, respectivamente) por efectos adversos, muerte o pérdida de seguimiento en la rama de EFV10.
Recomendaciones
- Se recomienda la administración de TAR a todos los pacientes con infección por el VIH-1 confirmada y tras valoración médica (A-I). Como potencial excepción se consideran los pacientes que mantienen CVP indetectable de forma mantenida sin TAR (controladores de élite).
- Se recomienda iniciar el TAR tan pronto como sea posible. En caso de iniciar el TAR antes de disponer de esos resultados, debe ser con un régimen que no precise el estudio previo de HLA-B*5701, con mínimo riesgo de interacciones farmacológicas, alta probabilidad de mantener actividad antiviral en escenarios “difíciles” (CVP elevadas, cifras bajas de linfocitos CD4+ o virus con mutaciones de resistencia basales) y capacidad de suprimir la replicación del VHB (A-II).
- Se debe realizar siempre una determinación de linfocitos CD4+ y CVP con estudio de resistencias previa al inicio del tratamiento, aunque no hay razón científica para esperar hasta disponer de los resultados si se utiliza una pauta cuya recomendación no esté condicionada a sus valores (A-III).
- Además, siempre debe prepararse al paciente, proporcionándole información sobre los objetivos del tratamiento y las distintas opciones, y valorando el riesgo de mala adherencia (A-III).
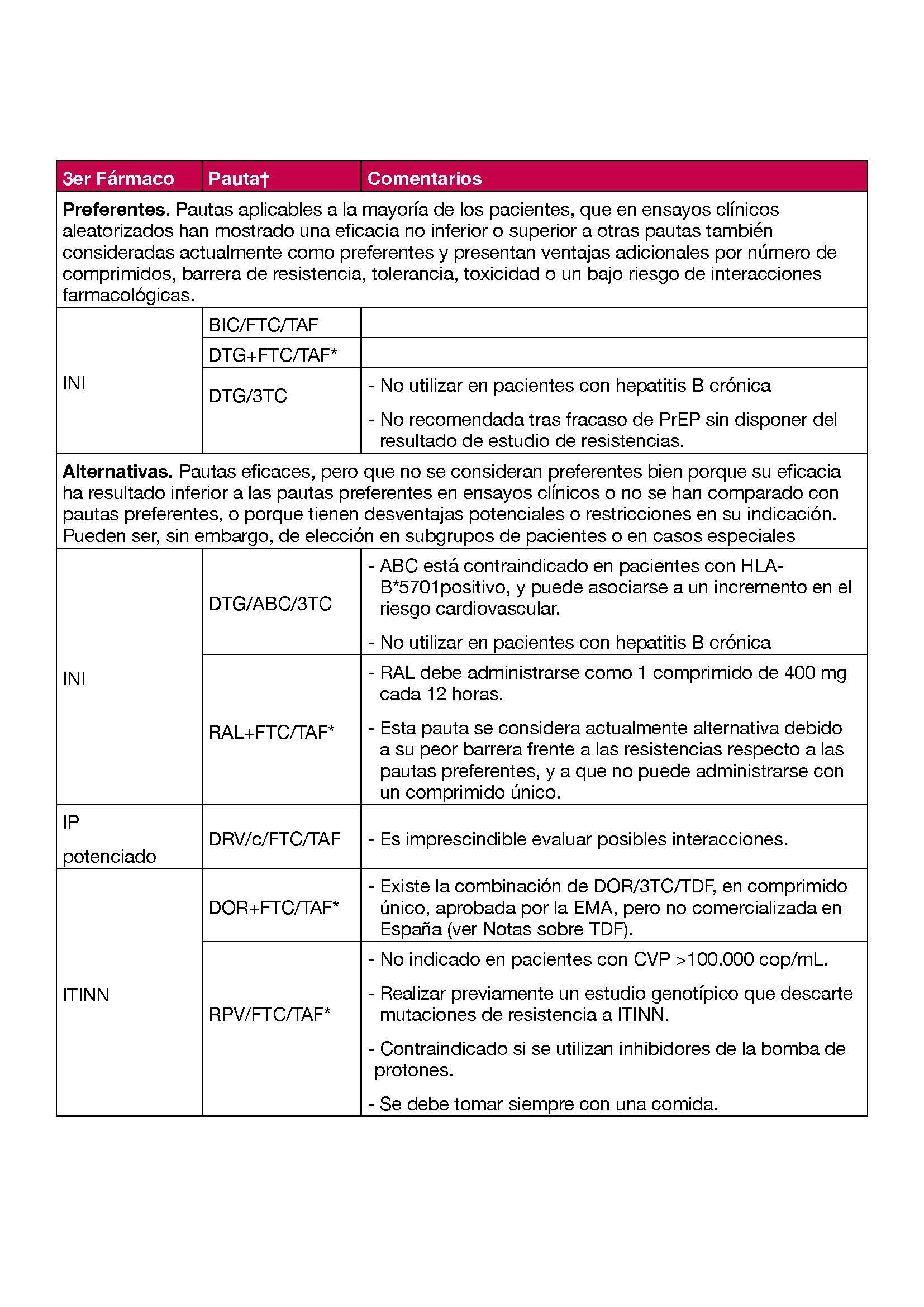
3.1.2. QUE COMBINACIÓN DE ANTIRRETROVIRALES DEBE UTILIZARSE
Las pautas recomendadas para el TAR inicial en el momento actual consisten en una combinación de dos o tres fármacos basados en un inhibidor de la integrasa (INI) de segunda generación (DTG o BIC) Tabla 2. La única pauta doble recomendada en el momento actual como TAR de inicio consiste en DTG/3TC. Estas combinaciones han demostrado en ensayos clínicos obtener una CVP <50 cop/mL a las 48 semanas de tratamiento en más del 85% de los casos.
En el caso de PVV embarazadas, con tuberculosis (TB), coinfección por VHB o virus de hepatitis C (VHC) o con historia de PrEP se debe utilizar la información existente en los apartados correspondientes de este documento y en las guías específicas.
Recomendación
- El TAR de inicio consiste en una combinación de dos o tres FAR, en alguna de las combinaciones que se detallan en la Tabla 2. (A-I)
3.1.2.1. INHIBIDORES DE LA TRANSCRIPTASA INVERSA ANÁLOGOS DE NUCLEÓSIDOS/NUCLEÓTIDOS (ITIAN)
Actualmente se utilizan tres ITIAN: 3TC, FTC y ABC. También se dispone de un análogo de nucleótido, tenofovir (TFV). A efectos prácticos, la abreviatura ITIAN en esta guía incluye también a TFV. Se considera como combinación de ITIAN de elección la formada por FTC/TAF, que debería administrase siempre que sea posible en régimen coformulado.
La utilización de TFV como tenofovir disoproxil (desarrollado clínicamente como fumarato, TDF) puede considerarse una alternativa a TAF, con reservas Tabla 2. En un metaanálisis sobre 14 ensayos clínicos (6 de ellos en pacientes naïve), se observó mayor eficacia y menos frecuentes discontinuaciones por toxicidad renal en los regímenes a base de TAF frente a TDF, cuando el régimen incluía un potenciador, sin diferencias de eficacia ni seguridad cuando el régimen no estaba potenciado11.
No existe evidencia clínica que permita afirmar diferente eficacia entre 3TC y FTC.
Combinaciones con FTC/TFV frente a combinaciones con ABC/3TC
Hasta este momento, TAF frente a ABC en el TAR de inicio sólo se ha comparado en el estudio GS-US-380-1489, aunque en regímenes diferentes (BIC/FTC/TAF vs DTG/ABC/3TC, doble ciego)12. En este estudio la eficacia de la combinación con TAF fue no inferior a la de ABC, aunque en el grupo que recibió la pauta con ABC la frecuencia de efectos adversos (EA) (náuseas) fue significativamente mayor.
Varios estudios han comparado TDF frente a ABC. El ensayo clínico ACTG 5202 comparó de forma ciega el inicio de TAR con ABC/3TC o FTC/TDF en 1.857 participantes, aleatorizados además a recibir ATV/r o EFV de forma abierta. Entre los pacientes con CVP basal igual o mayor de 100.000 cop/mL, tanto el tiempo hasta el FV como el tiempo hasta el primer efecto adverso de grado 3-4 fueron significativamente más cortos en el brazo de ABC/3TC que en el brazo de FTC/TDF. En los pacientes con CVP <100.000 cop/mL no hubo diferencias en eficacia virológica entre ABC/3TC y FTC/TDF, independientemente de que se administraran con ATV/r o EFV1314.
Tres estudios en fase III diseñados para comparar el TAR de inicio con DTG, frente a otros FAR recomendados (EFV en el estudio SINGLE15, raltegravir [RAL] en el estudio SPRING-216 o DRV/r en el estudio FLAMINGO17) han mostrado una eficacia similar de ABC/3TC o FTC/TDF, aunque la elección del ITIAN no fue aleatorizada.
Por otra parte, la prescripción de ABC requiere disponer de un HLA-B*5701 negativo y ABC no tiene actividad frente al VHB.
Estudios observacionales y metaanálisis sugieren, con datos discordantes, un posible aumento del riesgo de infarto de miocardio y de enfermedad cardiovascular en pacientes que recibieron ABC, especialmente con exposición reciente, por lo que su utilización debe evitarse en pacientes con riesgo cardiovascular elevado18.
El ensayo clínico aleatorizado REPRIEVE ha reportado una tasa significativamente mayor de eventos cardiovasculares en PVV tratados (previa o actualmente) con ABC19.
Por estos motivos, no se considera preferente el inicio de un TAR con ABC.
Recomendaciones
- La combinación de ITIAN recomendada para regímenes de inicio es FTC/TAF (A-I).
- TDF puede utilizarse como alternativa a TAF en regímenes que no incluyan un potenciador, ritonavir (RTV) o cobicistat (COBI), siempre que se excluya la presencia de alteración renal u ósea o aumento del riesgo de desarrollarlas. (C-I)
- La combinación ABC/3TC se recomienda como pauta alternativa de inicio (A-I), al tener peor tolerancia gastrointestinal, menor actividad contra VHB, requerir test HLA-B*5701 previo, la posible asociación de ABC con incremento del riesgo cardiovascular, y no haber demostrado ventajas sobre FTC/TAF (B-I).
3.1.2.2. INHIBIDORES DE LA INTEGRASA (INI)
No hay experiencia con CAB en tratamiento de inicio. El TAR de acción prolongada con CAB+RPV intramuscular (IM) solamente es aplicable como cambio en pacientes con viremia indetectable, alcanzada con alguno de los regímenes de inicio recomendados.
EVG, disponible en coformulación de EVG/c/FTC/TAF o TDF, no se incluye entre las pautas preferentes ni alternativas debido a su mayor potencial de interacciones con respecto a los otros INI no potenciados y su menor barrera a la resistencia.
DTG se ha comparado en ensayos clínicos fase III con FAR de las tres familias que en algún momento han sido recomendados en el TAR de inicio.
El ensayo clínico aleatorizado y doble ciego SINGLEdemostró la superioridad de DTG/3TC/ABC frente a EFV/FTC/TDF a las 48 semanas, que se mantuvo en la semana 144. La proporción de FV fue similar en ambos brazos de tratamiento, pero la proporción de interrupciones por EA fue mayor en el brazo de EFV/FTC/TDF.
En el estudio ADVANCE20, aleatorizado abierto, se incluyeron 1053 participantes naïve de Johannesburgo (99% de raza negra, 59% mujeres), tratados con DTG+FTC/TAF, DTG+FTC/TDF o EFV+FTC/TDF, demostrando la no inferioridad de DTG frente a EFV. Hubo más discontinuaciones en la rama de EFV, con más frecuente desarrollo de resistencias en los fracasos de esta rama, pero con mayor aumento de peso corporal en los grupos tratados con DTG+FTC/TAF, especialmente en las mujeres.
En el ensayo SPRING-216 aleatorizado y doble ciego, se incluyeron 822 pacientes naïve, que recibieron TAR con DTG o RAL 400 mg/12 horas, junto con dos ITIAN, demostrando la no inferioridad de DTG a las 48 semanas, aunque la eficacia virológica a las 96 semanas fue significativamente superior en la rama de DTG entre los participantes con viremia basal >100.000 cop/mL. No se desarrollaron MR y la tolerabilidad fue excelente en ambas ramas de tratamiento.
DTG ha demostrado eficacia superior a DRV/r, ambos en combinación con 2 ITIAN, a las 48 y a las 96 semanas, en un ensayo clínico aleatorizado abierto (FLAMINGO)17. Aunque no se observaron diferencias en cuanto a FV, el porcentaje de EA y discontinuaciones por causas no relacionadas con el fármaco fue mayor en los tratados con DRV/r.
Dos ensayos clínicos aleatorizados, doble ciego, con el mismo diseño (GEMINI-1 y GEMINI-2), han comparado la biterapia DTG+3TC con el TAR triple DTG+TDF/FTC en un total de 1.433 PVV sin tratamiento previo y con CVP <500.000 cop/mL. DTG+3TC demostró a las 48 semanas la no inferioridad frente al tratamiento triple (CVP <50 cop/mL en el 90% y 93% de los participantes, respectivamente, análisis snapshot; diferencia -1,7% (IC95% -4,4 a 1,1)21, que se mantuvo a las 96 semanas. En el análisis planificado de subgrupos, no se observaron diferencias en función de la CVP basal mayor o menor de 100.000 cop/mL. En cambio, los pacientes que iniciaron DTG+3TC con una cifra basal de CD4+ <200 células/μL mostraron una eficacia significativamente menor en el análisis snapshot, con respecto a los que recibieron terapia triple (CVP <50 cop/mL a 48 semanas en el 79% y 93% de los participantes, respectivamente, análisis snapshot; diferencia 13,4%), aunque esta menor eficacia no se debió a un mayor porcentaje de FV, sino a discontinuaciones no relacionadas directamente con el tratamiento. Además, el número de pacientes en este subgrupo, como sucede en la mayoría de los ensayos clínicos, fue pequeño (n=65, 9%) y no permite extraer conclusiones definitivas. En el análisis de los resultados a 144 semanas22, se mantuvo la no inferioridad de DTG+3TC, tanto global como en el análisis pormenorizado por subgrupos, y persistía la tendencia a una menor eficacia en los pacientes con una cifra basal de linfocitos CD4+ <200 células/μL (67% vs 76%, respectivamente; diferencia -9,7%; IC95% -25,9 a 6,5). Recientemente se han comunicado los resultados del ensayo clínico DOLCE, incluyendo 229 pacientes sin TAR previo, con recuentos de CD4+ ≤ 200 células/μL, aleatorizados a recibir DTG/3TC frente a DTG+TDF+(3TC o FTC), y se demuestran tasas de respuesta en el objetivo principal con DTG/3TC similares a la terapia triple (CVP <50 cop ARN/mL en el 82,2% y 80,5% de los pacientes, respectivamente, a las 48 semanas; análisis snapshot, diferencia 2%; IC95% -8,7 a 12,8). En un análisis post-hoc la respuesta virológica obtenida con ambos regímenes es superponible entre sí dentro de los distintos estratos de viremia, incluso por encima de 1.000.000 cop ARN/mL. Estos datos consolidan el posible uso de DTG/3TC como terapia de inicio, sin restricciones impuestas por la situación inmunovirológica de los pacientes 23.
En un metaanálisis en red de 14 ensayos clínicos aleatorizados y doble ciego con pautas de TAR de inicio se ha mostrado que la eficacia y seguridad de DTG+3TC a las 48 semanas de tratamiento es comparable a las combinaciones de TAR triples analizadas, incluso en aquellos pacientes con CVP basal >100.000 cop/mL24.
Es importante destacar que DTG presenta una alta barrera a las resistencias, siendo excepcional la selección de MR tras un FV, tanto en asociación con 2 ITIAN como con la biterapia DTG+3TC.
BIC se encuentra disponible en coformulación con FTC y TAF en un único comprimido que se administra en una pauta una vez al día (QD) (BIC/FTC/TAF).
BIC/FTC/TAF en pacientes sin TAR previo se ha estudiado en dos ensayos clínicos fase III aleatorizados y doble ciego, ambos en comparación con DTG. En el estudio GS-US-380-1489BIC/FTC/TAF demostró una eficacia no inferior a DTG/3TC/ABC y en el estudio GS-US-380-149025, BIC/FTC/TAF se mostró no inferior a DTG+FTC/TAF en ambos estudios a los 4 años. El porcentaje de discontinuaciones por EA fue bajo (2%) y similar a los grupos de tratamiento con DTG. No se observó selección de MR a BIC/FTC/TAF en ningún paciente, hasta 5 años de seguimiento26.
En el estudio ALLIANCE, aleatorizado en fase III, doble-ciego, en pacientes coinfectados por VIH y VHB, BIC/FTC/TAF ha demostrado la no inferioridad para la supresión del ARN del VIH-1 con respecto a DTG+FTC/TDF, y la superioridad para la supresión de los niveles de ADN del VHB a las 48 semanas, junto con tasas más elevadas de normalización de transaminasas y seroconversión del AgHBe, a 48 y 96 semanas27.
RAL no existe coformulado y presenta una baja barrera al desarrollo de resistencias. El estudio aleatorizado, doble ciego QDMRK en TAR de iniciocomparó la administración de RAL QD (n=382) vs BID (n=388). RAL QD no cumplió la no inferioridad en el objetivo primario (ARN < 50 cop/mL, a 48 semanas), fue inferior en PVV con ARN >100.000 cop/mL, y no cumplió la no inferioridad en PVV con CD4+ < 200 células/μL. Además, el riesgo de fracaso virológico fue mayor en PVV con CVP <100.000 cop/mL con RAL QD. El comité de seguridad recomendó a las 48 semanas detener el estudio.
Aunque en el ensayo clínico ONCEMRK la pauta con la nueva formulación de RAL 2 x 600 mg QD demostró ser no inferior al régimen BID, solo se observó el desarrollo de mutaciones de resistencias en algunos de los pacientes con FV que recibieron RAL QD29.
Recomendaciones
- Se recomiendan como pautas preferentes del TAR de inicio las siguientes combinaciones: BIC/FTC/TAF, DTG+FTC/TAF y DTG/3TC. (A-I)
- Si se considera el uso de una pauta alternativa basada en RAL en el TAR de inicio, este debe administrarse en régimen de 400 mg BID. (A-I)
3.1.2.3. INHIBIDORES DE LA TRANSCRIPTASA INVERSA NO NUCLEÓSIDOS (ITINN)
Actualmente no se recomienda el uso de NVP en pautas de TAR de inicio debido a su mayor riesgo de toxicidad y a no haber demostrado la no inferioridad con respecto a EFV o INI.
ETR no está aprobada por la EMA para el TAR de inicio.
EFV se ha comparado con INI en diversos ensayos clínicos aleatorizados de TAR de inicio que han puesto en evidencia una menor eficacia de EFV respecto a RAL (estudio STARTMRK30), DTG (estudio SINGLE15) o BIC10.
RPV se ha comparado con EFV en los estudios ECHO y THRIVE31, en que los participantes fueron aleatorizados a recibir de forma ciega RPV o EFV junto a dos ITIAN. El análisis combinado de ambos estudios a las 96 semanas demostró la no inferioridad de RPV con respecto a EFV, aunque en el subgrupo de pacientes con CVP inicial >100.000 cop/mL, la frecuencia de FV fue superior con RPV y se asoció con mayor frecuencia a resistencia genotípica a otros ITINN y a ITIAN (especialmente por la selección de las mutaciones M184I y M184V). La tolerabilidad fue mejor con RPV, con un menor número de discontinuaciones por EA.
En el ensayo clínico abierto STaR32 se compararon las combinaciones en un comprimido único de RPV/FTC/TDF frente a EFV/FTC/TDF como pautas de TAR de inicio. Se demostró la no inferioridad de RPV/FTC/TDF frente a EFV/FTC/TDF tanto a las 48 como a las 96 semanas. En el análisis de subgrupos la eficacia de RPV/FTC/TDF fue superior a EFV/FTC/TDF en los pacientes con CVP basal ≤100.000 cop/mL, no inferior en los pacientes con CVP >100.000 cop/mL e inferior en los pacientes con CVP >500.000 cop/mL. La retirada del tratamiento por EA, así como la incidencia de EA neuropsiquiátricos fue menor en los pacientes tratados con RPV en comparación con EFV.
La combinación RPV/FTC/TAF no ha sido evaluada de forma específica como TAR de inicio. Sin embargo, este panel considera que las ventajas en seguridad mostradas en las comparaciones directas de TAF con TDF en pacientes sin tratamiento previo, así como en los ensayos clínicos que han comparado la eficacia y seguridad del cambio a RPV/FTC/TAF en pacientes pretratados33 son suficientes para recomendar esta combinación como alternativa a las pautas preferentes.
DOR es un ITINN que se ha estudiado como tratamiento de inicio en ensayos clínicos aleatorizados comparándose con EFV, coformulados respectivamente con 3TC/TDF o con FTC/TDF (Estudio DRIVE-AHEAD)34, y con DRV/r, en combinación con FTC/TDF o 3TC/ABC (Estudio DRIVE-FORWARD)35. En ambos estudios, DOR ha mostrado una eficacia no inferior a sus comparadores en el análisis primario a las 48 semanas. El porcentaje de pacientes con EA sobre el SNC fue significativamente inferior en los tratados con DOR en comparación con EFV. En un análisis con 96 semanas de seguimiento ciego35, DOR ha demostrado eficacia superior a DRV/r con mejor evolución del perfil lipídico. El aumento de peso observado a las 96 semanas en los participantes de estos dos ensayos clínicos (junto con los de otro estudio fase IIb previo, de cálculo de dosis de DOR) fue similar con los regímenes a base de DOR, DRV/r o EFV36.
Hasta el momento DOR no se ha comparado con INI en ensayos clínicos.
Aunque no existe evidencia directa del uso de DOR con FTC/TAF, este Panel considera que las ventajas en seguridad mostradas en las comparaciones directas de TAF frente a TDF, junto con otros FAR, en pacientes sin tratamiento previo son suficientes para recomendar el uso de esta combinación como alternativa a las pautas preferentes.
Recomendaciones
- Actualmente no se considera preferente ninguna pauta basada en ITINN. (A-III)
- DOR+FTC/TAF o DOR+FTC/TDF se considera una alternativa a las pautas preferentes en el TAR de inicio. (C-I)
- En pacientes con CVP <100.000 cop/mL la combinación RPV/FTC/TAF se considera una alternativa a las pautas preferentes en el TAR de inicio. (C-I)
- RPV no debe utilizarse en pacientes con CVP >100.000 cop/mL. (A-I)
3.1.2.4. INHIBIDORES DE LA PROTEASA POTENCIADOS (IP/p)
En el TAR de inicio sólo se pueden usar IP potenciados con RTV o COBI. DRV/p es el único IP usado en el TAR inicial, bien sea potenciado con RTV, coformulado con COBI (DRV/c) o en un comprimido único con FTC/TAF (DRV/c/FTC/TAF).
El estudio ARTEMIS37 comparó DRV/r (800/100 mg, QD) frente a LPV/r en 689 pacientes que recibieron además FTC/TDF coformulados. A las 96 semanas, DRV/r resultó superior a LPV/r (TLOVR) y abandonaron el tratamiento 4% y 9% de los pacientes tratados con DRV/r y LPV/r, respectivamente.
DRV/r se comparó con RAL en el estudio ACTG 525738 sin objetivar diferencias en el porcentaje de FV. En el análisis snapshot y en el análisis conjunto de la respuesta virológica y tolerabilidad DRV/r fue inferior a RAL.
DRV/r también se mostró inferior a DTG en el ensayo clínico FLAMINGO17, debido fundamentalmente a una mayor tasa de EA y discontinuaciones por causas no relacionadas con el fármaco.
En el estudio abierto SYMTRI39 se aleatorizaron 316 pacientes naïve, para recibir DRV/c/FTC/TAF o DTG/ABC/3TC. Tras 48 semanas, DRV/c/FTC/TAF tuvo similares resultados de eficacia que DTG/ABC/3TC, pero no alcanzó la no inferioridad.
En el ensayo clínico aleatorizado y doble ciego AMBER se comparó DRV/c administrado junto con FTC/TDF o coformulado con FTC/TAF en comprimido único, en PVV sin tratamiento previo40 y con CD4+ >50 células/μL. La combinación DRV/c/FTC/TAF demostró una eficacia no inferior a las 48 semanas.
Debido al mayor riesgo de interacciones farmacológicas y no haber demostrado la no inferioridad respecto a las combinaciones de TAR consideradas preferentes, las combinaciones con DRV/p sólo se recomiendan como alternativas en pautas de inicio.
Recomendaciones
- Actualmente no se considera preferente para TAR de inicio ninguna pauta basada en IP/p. (A-III)
- Cuando se considere conveniente iniciar un tratamiento basado en IP se recomienda utilizar DRV/c/FTC/TAF (A-I).
SITUACIONES ESPECIALES
3.2.1. INFECCIÓN AGUDA POR EL VIH
Se considera infección reciente al periodo ≤6 meses en el que se detectan anticuerpos frente al VIH-1, e infección aguda o primoinfección al periodo de aproximadamente 30-60 días tras la infección en el que se alcanza una estabilización de la viremia41. La primoinfección se caracteriza por síntomas inespecíficos compatibles con cualquier otra infección viral (fiebre, cefalea, inflamación de ganglios linfáticos, erupciones cutáneas y/o malestar general) o con síntomas muy leves, o por ser asintomática, pero también con cuadros más graves como meningoencefalitis41. Por tanto, su diagnóstico se basa en la detección del virus en plasma (ARN del VIH-1 y/o antígeno p24) y/o evolución de la reactividad de anticuerpos frente al VIH-1, de negativo o indeterminado a positivo.
El TAR precoz en la primoinfección acorta la duración y gravedad de los síntomas, suprime rápidamente la replicación viral, reduce la diversidad viral y el reservorio VIH-1, normaliza más rápidamente la cifra de linfocitos T CD4+ y el cociente CD4/CD8, reduce la activación inmunológica, preserva o restaura la inmunidad específica frente al VIH-1 y reduce la transmisión424344. Pese a que existe un grupo de personas que tratadas precozmente (<90 días) consiguen controlar el virus a niveles indetectables en plasma tras la interrupción de tratamiento (controladores post-tratamiento)45, no se han encontrado parámetros capaces de predecir con robustez el rebote viral tras la interrupción, y por tanto, no se recomienda.
Recomendaciones
- Se recomienda en todos los casos una prueba de resistencias lo antes posible después del diagnóstico, cuyo resultado no tiene que retrasar el inicio del TAR (A-III).
- Se recomienda iniciar el TAR en todos los casos, tan pronto como sea posible (A-II) y mantener indefinidamente (A-I).
- El tipo de TAR será el mismo que en la infección crónica, basado en un INI de alta barrera genética (DTG o BIC) con TDF o TAF + 3TC o FTC. (A-I). Si es posible, debe ofrecerse la participación en un ensayo o estudio clínico que investigue estrategias curativas del VIH (B-III).
3.2.2. INFECCIÓN POR EL VIH EN SUJETO EN PREP
La infección aguda por el VIH en usuarios de PrEP es infrecuente, no obstante, el riesgo de desarrollar MR al TAR utilizado (actualmente TDF/FTC) aumenta, especialmente cuando se adquiere la infección justo antes de iniciar PrEP o en sujetos con adherencia muy irregular, pero que no la hayan suspendido definitivamente4647. En este escenario, la seroconversión podría ser más tardía y la CVP menor, llevando a resultados ambiguos que dificultan el diagnóstico4849.
Las tasas de selección de resistencia varían según la tasa de infección por VIH en período ventana al inicio de PrEP y la adherencia. Debe sospecharse la presencia de M184V/I (el 23% de las personas con test de resistencia disponible en una revisión reciente presentaban esta mutación), y mucho más raramente K65R (1,3% de los participantes)46.
Aunque en España aún no está aprobado su uso, el empleo de CAB de acción prolongada como PrEP, altera la clínica habitual de infección aguda por VIH y las pruebas serológicas pueden no ser útiles para el diagnóstico temprano, con riesgo de desarrollo de mutaciones emergentes a INIs50. La CVP debe repetirse, para descartar posibles falsos negativos51.
Recomendaciones
- Realizar un test genotípico de resistencias en VIH antes de iniciar el TAR (A-II).
- Si se sospecha infección aguda, además de una serología ELISA de 4ª generación, si los resultados son confusos o existe una alta sospecha, debe repetirse la serología en una nueva muestra y solicitar CVP (A-II).
- Iniciar el TAR siempre con triple terapia con fármacos de alta barrera a la resistencia (TDF ó TAF/FTC + DTG o BIC o DRV/c) en espera del test genotípico (A-III).
- En pacientes que hayan recibido CAB de acción prolongada como PrEP debe solicitarse siempre CVP de forma repetida como estrategia de cribado de infección por VIH y el TAR de elección es FTC/TDF + DRV/c o DRV/c/FTC/TAF (A-II).
3.2.3. PACIENTES CON EVENTO OPORTUNISTA
El TAR debe iniciarse lo antes posible en PVV con una infección oportunista (IO). El ensayo clínico aleatorizado ACTG A5164 (n=282, excluyendo tuberculosis [TB]), demostró una menor tasa de progresión a SIDA o muerte (HR 0,53; IC95% 0,30 a 0,92) en PVV que iniciaban TAR dentro de una mediana de 12 días tras el inicio del tratamiento de la IO en comparación con aquellos que demoraban el inicio hasta una mediana de 45 días. No se observaron diferencias en la incidencia de efectos adversos ni de síndrome inflamatorio de reconstitución inmune (SIRI). Más del 60% de los pacientes presentaban neumonía por Pneumocystis jirovecii (NPJ). Dado el pequeño número y la variedad de otras IO, es difícil extraer conclusiones para cada una de ellas por separado52.
Existen dos escenarios clínicos en los que el inicio del TAR es motivo de controversia: la meningitis criptocócica y la tuberculosa. En una revisión sistemática de pacientes con meningitis criptocócica realizada por la colaboración Cochrane (4 estudios, 294 pacientes), se observó una mayor mortalidad cuando se iniciaba TAR de forma precoz (< 4 semanas desde el inicio del tratamiento antifúngico) vs tardío (RR 1,42; IC95% 1,02 a 1,97), sin asociarse a un aumento de SIRI53. En un subanálisis más reciente del estudio AMBITION-cm, diseñado para estudiar la eficacia de un régimen basado en una única dosis de anfotericina B liposomal, también se observó un exceso de mortalidad a las 2 y 10 semanas entre las personas que habían comenzado TAR dentro de los 14 días previos54. Sin embargo, una revisión retrospectiva de 190 PVV en países desarrollados (Europa y Norteamérica), en la que se garantiza el adecuado manejo de la hipertensión intracraneal secundaria, no ha encontrado diferencias en mortalidad entre el inicio diferido o precoz del TAR (RR 1,28 [IC95% 0,64 a 2,56] y 1,40 [IC95% 0,66 a 2,95], respectivamente)55. Es probable que parte de las diferencias con estudios realizados en países en desarrollo se expliquen por un manejo subóptimo de la hipertensión intracraneal en estos, sin embargo, el diseño retrospectivo del estudio, así como la escasez de datos con respecto al manejo de los pacientes impiden generalizar sus conclusiones.
Con respecto a la meningitis tuberculosa, se recomienda consultar el apartado específico más adelante en este documento.
No existen ensayos clínicos que comparen pautas de TAR en pacientes con IO, por lo que la selección del régimen debe seguir los mismos criterios que en pacientes sin IO, considerando especialmente las potenciales interacciones medicamentosas y la nefrotoxicidad.
Recomendaciones
- En PVV con IO se debe iniciar el TAR lo antes posible (dentro de las dos primeras semanas tras el inicio del tratamiento de la IO) (A-II), especialmente en pacientes con neumonía por Pneumocystis jirovecii (NPJ) (A-I).
- En pacientes con meningitis criptocócica se recomienda diferir el inicio del TAR entre 4 y 6 semanas (A-I) y garantizar un manejo adecuado de la hipertensión intracraneal.
- Sin considerar la TB, la pauta de tratamiento antirretroviral inicial empleada en pacientes con IO no tiene por qué diferir de las PVV sin IO (A-III).
Tablas:

Notas:
- Se consideran como potencial excepción los pacientes que mantienen CVP indetectable de forma mantenida sin TAR (controladores de élite). En este caso no existe información que permita valorar el efecto beneficioso del TAR, por lo que no se puede establecer una recomendación al respecto.
- La disposición y la motivación del paciente es un factor crítico a la hora de tomar la decisión de empezarlo. Es importante hacer una valoración individualizada del momento de inicio del TAR y de los FAR que deben formar parte del régimen inicial, sopesando las ventajas e inconvenientes de cada una de las opciones.
† En el caso de personas embarazadas, con tuberculosis, coinfección por el VHB o VHC, o con historia de
PrEP estas recomendaciones no son válidas y se debe utilizar la información existente en los apartados
correspondientes y las guías específicas.
† Si se opta por un inicio rápido tras el diagnóstico, es habitual no disponer del resultado del estudio de
resistencias ni de la determinación de HLA-B*5701, por lo que no se deben utilizar regímenes basados en ITINN ni con ABC. Si se inicia el TAR antes de disponer de los resultados del recuento de linfocitos CD4+ o CVP hay que evitar de inicio los regímenes cuya recomendación esté condicionada por estos resultados (como los basados en RPV).
† Cuando estén disponibles, se recomienda el uso de preparados que combinen fármacos a dosis fijas. Los ensayos clínicos en los que se fundamenta la evidencia de cada pauta se referencian en el texto.
† Los comentarios reflejan aspectos que se deben considerar en la elección de régimen, pero no pretenden ser una guía exhaustiva de las precauciones a tomar en el uso de los fármacos. Para mayor información se recomienda revisar el texto del documento, así como las fichas técnicas de los fármacos.
* La utilización de TFV como TDx (fundamentalmente TDF) puede considerarse una alternativa a TAF cuando no se asocie a un fármaco potenciado y siempre que se excluya la presencia de alteración renal o de osteopenia/osteoporosis, y no existan otros factores de riesgo para desarrollarlas. Por otra parte, TDF puede administrarse coformulado con FTC o con 3TC.
Bibliografía:
Lundgren JD, Babiker AG, Gordin F, et al. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med 2015; 373:795-807.
Danel C, Moh R, Gabillard D, et al. A trial of early antiretrovirals and isoniazid preventive therapy in Africa. N Engl J Med 2015; 373:808-22.
Cohen MS, Chen YQ, McCauley M, et al. Antiretroviral therapy for the prevention of HIV-1 transmission. N Engl J Med 2016; 375:830-9.
Ford N, Migone C, Calmy A, et al. Benefits and risks of rapid initiation of antiretroviral therapy. AIDS 2018; 32:17-23.
Huhn GD, Crofoot G, Ramgopal M, et al. Darunavir/Cobicistat/Emtricitabine/Tenofovir Alafenamide in a rapid-initiation model of care for human immunodeficiency virus type 1 infection: primary analysis of the DIAMOND Study. Clin Infect Dis 2020; 71:3110-7.
Rolle C-P, Berhe M, Singh T, et al. Sustained Virologic Suppression with Dolutegravir/
Bachelard A, Isernia V, Charpentier C, et al. Same-day initiation of bictegravir/emtricitabine/ tenofovir alafenamide: Week 48 results of the FAST study—IMEA 055. J Amtimicrob Chemother 2023; 78:769-78.
Hidalgo-Tenorio C, Sequera S, Vivancos MJ, et al. Bictegravir/emtricitabine/tenofovir alafenamide as first-line treatment in naïve HIV patients in a rapid-initiation model of care: BIC-NOW clinical trial. Int J Antimicrob Agents 2024;63(6):107164. https://doi.org/10.1016/j.ijantimicag.2024.107164.
Ghosn J, Chow J, Gandhi M, et al. Rapid start with bictegravir/emtricitabine/tenofovir alafenamide (B/F/TAF) as initial treatment in people with HIV-1 (PWH): A systematic literature review (SLR) of clinical and patient-reported outcomes (PROs). HIV Drug Therapy. Glasgow 2024, P154.
Wang R, Sun L, Wang X, et al. Rapid initiation of antiretroviral therapy with coformulated bictegravir, emtricitabine, and tenofovir alafenamide versus efavirenz, lamivudine, and tenofovir disoproxil fumarate in human immunodeficiency virus–positive men who have sex with men in China: Week 48 results of the multicentre, randomized clinical trial. Clin Infect Dis 2024; https://doi.org/10.1093/cid/ciae012.
Pilkington V, Hughes SL, Pepperrell T, et al. Tenofovir alafenamide vs. tenofovir disoproxil fumarate: an updated meta-analysis of 14 894 patients across 14 trials. AIDS 2020; 34:2259-68.
Gallant J, Lazzarin A, Mills A, et al. Bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir, abacavir, and lamivudine for initial treatment of HIV-1 infection (GS-US-380-1489): a double-blind, multicentre, phase 3, randomised controlled non-inferiority trial. Lancet 2017; 390:2063-72.
Sax PE, Tierney C, Collier AC, et al. Abacavir/lamivudine versus tenofovir DF/emtricitabine as part of combination regimens for initial treatment of HIV: final results. J Infect Dis 2011; 204:1191-201.
Daar ES, Tierney C, Fischl MA, et al. Atazanavir plus ritonavir or efavirenz as part of a 3-drug regimen for initial treatment of HIV-1. Ann Intern Med 2011; 154:445-56.
Walmsley S, Baumgarten A, Berenguer J, et al. Dolutegravir plus abacavir/lamivudine for the treatment of HIV-1 infection in antiretroviral therapy-naive patients: week 96 and week 144 results from the SINGLE randomized clinical trial. J Acquir Immune Defic Syndr 2015; 70:515-9.
Raffi F, Jaeger H, Quirós-Roldán E, et al. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial. Lancet Infect Dis 2013; 13:927-35.
Molina JM, Clotet B, van Lunzen J, et al. Once-daily dolutegravir versus darunavir plus ritonavir for treatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomised, open- label, phase 3b study. Lancet HIV 2015; 2: e127-36.
Dorjee K, Choden T, Baxi SM, et al. Risk of cardiovascular disease associated with exposure to abacavir among individuals with HIV: A systematic review and meta-analyses of results from 17 epidemiologic studies. Int J Antimicrob Agents 2018; 52:541-553.
Fichtenbaum CJ, Malvestutto CD, Watanabe MG, et al. Abacavir is associated with elevated risk for cardiovascular events in the REPRIEVE trial. IAS AIDS 2024 July 22-26 Munich. Abstr OAB3406LB.
Venter WDF, Moorhouse M, Sokhela S, et al. Dolutegravir plus Two Different Prodrugs of Tenofovir to Treat HIV. N Engl J Med 2019;381:803-15.
Cahn P, Sierra Madero J, Arribas JR, et al. Dolutegravir plus lamivudine versus dolutegravir plus tenofovir disoproxil fumarate and emtricitabine in antiretroviral-naive adults with HIV-1 infection (GEMINI-1 and GEMINI-2): week 48 results from two multicentre, double-blind, randomised, non-inferiority, phase 3 trials. Lancet 2019; 393:143-55.
Cahn P, Sierra Madero J, Arribas JR, et al. Three-year durable efficacy of dolutegravir plus lamivudine in antiretroviral therapy-naïve adults with HIV-1 infection. AIDS 2022; 36:39-48.
Figueroa MI, Brites C, Cecchini D, et al. Comparable efficacy and safety of dolutegravir / lamivudine to a three drug regimen amongst ARV naive people living with HIV with CD4 <200/mm3: The DOLCE study. HIV Drug Therapy, Glasgow 2024. O24.
Radford M, Parks DC, Ferrante S, Punekar Y. Comparative efficacy and safety and dolutegravir and lamivudine in treatment naive HIV patients. AIDS 2019; 33:1739-49.
Sax PE, Pozniak A, Montes ML, et al. Coformulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection (GS-US-380-1490): a randomised, double-blind, multicentre, phase 3, non-inferiority trial. Lancet 2017; 390:2073-82.
Sax PE, Arribas JR, Orkin C, et al. Bictegravir/emtricitabine/tenofovir alafenamide as initial treatment for HIV-1: five-year follow-up from two randomized trials. eClinicalMedicine 2023; 59:101991.
Avihingsanon A, Lu H, Leong CL, et al. Bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir, emtricitabine, and tenofovir disoproxil fumarate for initial treatment of HIV-1 and hepatitis B coinfection (ALLIANCE): a double-blind, multicentre, randomised controlled, phase 3 non-inferiority trial. Lancet HIV. 2023 Oct;10: e640-e652. doi: 10.1016/S2352-3018(23)00151-0. Epub 2023 Jul 23.
Eron JJ Jr, Rockstroh JK, Reynes J, et al. Raltegravir once daily or twice daily in previously untreated patients with HIV-1: a randomised, active-controlled, phase 3 non-inferiority trial. Lancet Infect Dis 2011; 11:907–15
Cahn P, Kaplan R, Sax PE, et al. Raltegravir 1200 mg once daily versus raltegravir 400 mg twice daily, with tenofovir disoproxil fumarate and emtricitabine, for previously untreated HIV-1 infection: a randomised, double-blind, parallel-group, phase 3, non-inferiority trial. Lancet HIV 2017; 4: e486–94.
Rockstroh JK, DeJesus E, Lennox JL, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naive HIV-1-infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr 2013; 63:77-85.
Nelson MR, Elion RA, Cohen CJ, et al. Rilpivirine versus efavirenz in HIV-1-infected subjects receiving emtricitabine/tenofovir DF: pooled 96-week data from ECHO and THRIVE Studies. HIV Clin Trials 2013; 14:81-91.
Cohen C, Wohl D, Arribas JR, et al. Week 48 results from a randomized clinical trial of rilpivirine/ emtricitabine/tenofovir disoproxil fumarate vs. efavirenz/emtricitabine/tenofovir disoproxil fumarate in treatment-naive HIV-1-infected adults. AIDS 2014; 28:989-97.
Hagins D, Orkin C, Daar ES, et al. Switching to coformulated rilpivirine (RPV), emtricitabine (FTC) and tenofovir alafenamide from either RPV,FTC and tenofovir disoproxil fumarate (TDF) or efavirenz, FTC and TDF: 96-week results from two randomized clinical trials. HIV Med 2018; 19:724-33.
Orkin C, Squires KE, Molina JM, et al. Doravirine/Lamivudine/Tenofovir Disoproxil Fumarate is Non- inferior to Efavirenz/Emtricitabine/ Tenofovir Disoproxil Fumarate in Treatment-naive Adults with Human Immunodeficiency Virus-1 Infection: Week 48 Results of the DRIVE-AHEAD Trial. Clin Infect Dis 2019; 68:535-44.
Molina JM, Squires K, Sax PE, et al. Doravirine versus ritonavir-boosted darunavir in antirretroviral-naive adults with HIV-1 (DRIVE-FORWARD): 96-week results of a randomized, double-blind, non-inferiority, phase 3 trial. Lancet HIV 2020; 7: e16-e26.
Orkin C, Elion R, Thompson M, et al. Changes in weight and BMI with first-line doravirine based therapy. AIDS 2021; 35:91-9.
Orkin C, DeJesus E, Khanlou H, et al. Final 192-week efficacy and safety of once-daily darunavir/ ritonavir compared with lopinavir/ritonavir in HIV-1-infected treatment-naive patients in the ARTEMIS trial. HIV Med 2013; 14:49-59.
Lennox JL, Landovitz RJ, Ribaudo HJ, et al. Efficacy and tolerability of 3 nonnucleoside reverse transcriptase inhibitor-sparing antiretroviral regimens for treatment-naive volunteers infected with HIV- 1: A randomized, controlled equivalence trial. Ann Intern Med 2014; 161:461-71.
Podzamczer D, Micán R, Tiraboschi J, et al. Darunavir/Cobicistat/Emtricitabine/Tenofovir Alafenamide versus Dolutegravir/Abacavir/Lamivudine in antiretroviral-naive adults (SYMTRI): a multicenter randomized open-label study (PReEC/RIS-57). Open Forum Infect Dis 2022 Mar; 9(3): ofab595. https://doi.org/10.1093/ofid/ofab595.
Eron JJ, Orkin C, Gallant J, et al. A week-48 randomized phase-3 trial of darunavir/cobicistat/emtricitabine/tenofovir alafenamide in treatment-naive HIV-1 patients. AIDS 2018; 32:1431-42.
Robb ML, Eller LA, Kibuuka H, et al. Prospective study of acute HIV-1 infection in adults in east Africa and Thailand. N Engl J Med 2016;374(22):2120-2130. Available at: https://pubmed.ncbi.nlm.nih.gov/27192360.
Le T, Wright EJ, Smith DM, et al. Enhanced CD4+ T-cell recovery with earlier HIV-1 antiretroviral therapy. N Engl J Med. 2013 Jan 17;368(3):218-30.
Lama JR, Ignacio RAB, Alfaro R, Ríos J, Cartagena JG, Valdez R, Bain C, et Clinical and immunologic outcomes after immediate or deferred antiretroviral therapy initiation during primary human immunodeficiency virus infection: The Sabes Randomized Clinical Study. Clin Infect Dis. 2021 Mar 15;72(6):1042-1050.
Ambrosioni J, de Lazzari E, Suanzes P, et al. Bictegravir/emtricitabine/tenofovir alafenamide (BIC/FTC/TAF) for the treatment of primary HIV infection (PHI): the BIC-PHI clinical trial. IAS 2024 Poster_WEPEB098.
Sáez-Cirión A, Bacchus C, Hocqueloux L, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Patholog. 2013 Mar;9(3):e1003211.
Landovitz RJ, TaoL, Yang J, et al. Type 1 Human Immunodeficiency Virus (HIV-1) Incidence, Adherence, and Drug Resistance in Individuals Taking Daily Emtricitabine/Tenofovir Disoproxil Fumarate for HIV-1 Pre-exposure Prophylaxis: Pooled Analysis From 72 Global Sstudies. Clin Infect Dis. 2024 Nov 22;79(5):1197-1207.https://doi.org/10.1093/cid/ciae143.
Ambrosioni J, Petit E, Liegeon G, Laguno M, Miró JM. Primary HIV-1 infection in users of pre-exposure prophylaxis. Lancet HIV 2021; 8: e166-e174.
Donnell D, Ramos E, Celum C, et al. Partners PrEP Study Team. The effect of oral preexposure prophylaxis on the progression of HIV-1 seroconversion. AIDS 2017 10; 31: 2007-16.
Smith DK, Switzer WM, Peters P, et al. A strategy for PrEP clinicians to manage ambiguous HIV test results during follow-up visits. Open Forum Infect Dis 2018;5: ofy180.
Marzinke MA, Fogel JM, Wang Z, et al. Extended Analysis of HIV Infection in Cisgender Men and Transgender Women Who Have Sex with Men Receiving Injectable Cabotegravir for HIV Prevention: HPTN 083. Antimicrob Agents Chemother. 2023 Apr 18;67(4):e0005323.
RJ Landovitz, et al. Performance characteristics of HIV RNA screening with long-acting injectable cabotegravir (CAB-LA) pre-exposure prophylaxis (PrEP) in HPTN. International AIDS Conference. Tuesday, July 23, 2024.
Zolopa A, Andersen J, Powderly W, et al. Early antiretroviral therapy reduces AIDS progression/death in individuals with acute opportunistic infections: a multicenter randomized strategy trial. PLoS One 2009; 4: e5575. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0005575
Eshun-Wilson I, Okwen MP, Richardson M, Bicanic T. Early versus delayed antiretroviral treatment in HIV-positive people with cryptococcal meningitis. Cochrane Database Syst Rev 2018; 7:CD009012. https://doi.org/10.1002/14651858.CD009012.pub3
Moyo M, Lawrence DS, Kalata N, et al. Recent ART Initiation and Mortality Risk in HIV-Associated Cryptococcal Meningitis. [poster]. Presented at Conference of Retroviruses and Opportunistic Infections 2024, CO March 2-42024, Denver. #886.
Ingle SM, Miro JM, May MT, et al. Early Antiretroviral Therapy Not Associated with Higher Cryptococcal Meningitis Mortality in People with Human Immunodeficiency Virus in High-Income Countries: An International Collaborative Cohort Study. Clin Infect Dis. 2023 Jul 5;77(1):64-73. doi: 10.1093/cid/ciad122.
4. CAMBIO DEL TAR EN PACIENTES CON REPLICACIÓN VIRAL SUPRIMIDA
CONSIDERACIONES GENERALES
Las causas más comunes para el cambio del TAR en PVV con CVP suprimida son: 1) la simplificación del tratamiento, en cuanto al número de comprimidos diarios, número de fármacos o la frecuencia de la dosis; 2) la prevención y el control de toxicidades o comorbilidades; 3) las interacciones medicamentosas, potenciales o presentes y 4) el cambio de la vía de administración y 5) otras cuestiones relacionadas con la conveniencia y la calidad de vida1.
La nueva pauta de TAR debe mantener la supresión virológica y no comprometer opciones de tratamiento futuras.
En aquellas personas con coinfección por VHB, la retirada de fármacos con actividad también frente al VHB podría ocasionar un rebote viral del VHB con consecuencias potencialmente graves.2
En la Tabla 1 se resumen las recomendaciones sobre modificaciones del TAR en PVV con CVP suprimida.
Recomendaciones
- Antes del cambio de TAR debe revisarse la historia clínica y farmacológica por si influyeran en la selección de los nuevos fármacos, principalmente los cambios previos y sus motivos (intolerancia, toxicidad o fracaso virológico), además de los resultados de las pruebas de resistencia (A-III).
- Debe descartarse infección por VHB y revisarse la situación serológica antes de cambiar a un TAR sin TFV más 3TC o FTC. Las personas con infección crónica por VHB deberán mantener un tratamiento con antivirales eficaces frente al VHB en caso de cambios en el TAR (A-II).
- Tras el cambio de TAR es recomendable evaluar, en un plazo de 4 semanas, el mantenimiento de la supresión virológica, la tolerancia a la nueva pauta y, en su caso, la mejoría o desaparición de toxicidades previas (B-III).
INDICACIONES DE CAMBIO Y PAUTAS RECOMENDADAS
4.2.1 SIMPLIFICACIÓN
Se considera simplificación del TAR la reducción del número de dosis, de comprimidos o de fármacos, el cambio a una pauta sin requerimientos específicos para las tomas (por ejemplo, con alimentos) o el cambio a una pauta que requiera una menor necesidad de monitorizar parámetros de toxicidad. La simplificación del TAR tiene como objetivo mejorar la adherencia, la comodidad de las tomas y la calidad de vida, manteniendo la eficacia. En algunos casos, la simplificación puede suponer, además, una reducción del coste del TAR.
En PVV sin historia de FV o evidencia de MR las opciones de cambio son muy variadas, pudiendo utilizarse tanto aquellas que han demostrado su eficacia en personas naïve como las estudiadas específicamente en el escenario de cambio de tratamiento.
En este escenario, múltiples ensayos clínicos han demostrado la eficacia y seguridad de cambios de fármacos dentro de una misma clase de ARV, sustituciones por otros de diferentes clases, cambios a un comprimido diario o cambios a pautas con solo dos fármacos. Actualmente existe la opción de cambio de una pauta oral a una pauta por vía IM administrada cada 2 meses.
La combinación de 2 ITIAN + el INI de segunda generación BIC en formulación a dosis fija en un solo comprimido diario (BIC/FTC/TAF) es una opción segura y eficaz en un gran número de casos. Permite la simplificación tanto desde otras pautas con INI como desde regímenes basados en ITINN o IP/p. 345
Las pautas de TAR duales con DTG/3TC o DTG/RPV son, asimismo, opciones de simplificación para PVV tratadas con IP/p, ITINN o INI + 2 ITIAN sin historia de FV previo, evidencia o sospecha de resistencia a los fármacos de la pauta o presencia de infección activa por VHB. 678
Otras opciones que también han demostrado ser seguras y eficaces en simplificación son las combinaciones en comprimido único de 2 ITIAN con ITINN (RPV/FTC/TAF o DOR/3TC/TDF) o IP/p (DRV/c/FTC/TAF). 91011.
Otra alternativa en simplificación más reciente es el cambio a un TAR dual con CAB+RPV administrados por vía IM cada 2 meses. 12
En PVV con antecedentes de FV y/o MR a ITIAN se ha demostrado en EC aleatorizados que los cambios a las combinaciones BIC/FTC/TAF, DTG+FTC/TAF o DRV/c/FTC/TAF, pueden mantener la eficacia virológica. 10,13-16
Un EC reciente en PVV con antecedentes de detección de la mutación M184V y supresión de la CVP durante un tiempo prolongado ha mostrado que el cambio a DTG/3TC puede mantener la supresión viral, independientemente de que esta mutación pueda detectarse en ADN proviral mediante NGS.17
La combinación de BIC/FTC/TAF + DRV/c18, o la pauta de dos fármacos DTG + DRV/p 19también podrían ser opciones de simplificación de pautas más complejas en PVV con antecedentes de múltiples FV y MR, siempre que se mantenga preservada la actividad de DRV y los INI.
Recomendaciones
- En PVV sin MR y/o FV previos se recomienda utilizar las pautas más sencillas y cómodas. Las simplificaciones a los regímenes orales de una toma diaria BIC/FTC/TAF, DTG/3TC o DTG/RPV o la pauta CAB+RPV IM cada 2 meses son seguras y eficaces (A-I).
- En PVV con antecedentes de FV y MR o sin información sobre resistencias, la nueva pauta debe tener una barrera a la resistencia similar a la pauta supresora previa (A-III).
- En aquellas PVV con antecedente de MR a ITIAN no se recomienda el cambio a pautas con 2 ITIAN y un tercer fármaco de baja barrera genética como un ITINN o RAL (A-I).
- En PVV con antecedente de MR a ITIAN se puede considerar la simplificación a BIC/FTC/TAF, DTG+FTC/TAF o DRV/c/FTC/TAF, de forma individualizada y asegurando que la nueva pauta contiene al menos 2 fármacos activos (A-I).
- En PVV con pautas complejas debido a fracasos previos y MR, con actividad preservada a DRV, DTG y/o BIC, se puede simplificar a pautas basadas en estos fármacos con o sin ITIAN (A-I).
4.2.2 Prevención de toxicidades
A pesar del excelente perfil de seguridad y tolerancia de los nuevos ARVs, sigue habiendo EA potenciales (inmediatos o tardíos) que pueden anticiparse en función de las condiciones del paciente (comorbilidades o hábitos de vida) y de los propios ARVs recibidos. A la hora de plantear un cambio preventivo del TAR en una persona con la CVP suprimida Tabla 1 , se deben tener en cuenta el nivel de la evidencia, el historial de tratamiento, las características individuales de la persona, así como sus comorbilidades, la potencial toxicidad del TAR actual y de las pautas alternativas.
Hay evidencia procedente de EC aleatorizados de mejora de parámetros óseos y renales de pacientes que cambian de TDF a TAF o ABC20, de mejora en el perfil lipídico y de síntomas neuropsiquiátricos en pacientes que cambian de EFV a RPVo DOR10 y de la dislipidemia asociada a IP/p tras el cambio a RPV, DOR, RAL, DTG o BIC 4, 9-10, 22-23. En relación con los regímenes duales, el cambio a DTG/RPV desde pautas basadas en IP/p, ITINN o INI + 2 ITIAN que incluían TDF ha demostrado mejor evolución de parámetros de metabolismo óseo y densidad mineral ósea (DMO) 8 y el cambio a DTG/3TC desde combinaciones que incluían TAF ha demostrado mejor evolución de parámetros del perfil lipídico, especialmente en aquellas personas cuyo tratamiento contenía además COBI o RTV como potenciador.24
Se ha observado un mayor aumento de peso en PVV que cambian a un TAR con INI de segunda generación, especialmente en combinación con TAF, y de forma más acentuada en aquellas personas en las que se retira TDF y/o EFV 25. En un EC aleatorizado en PVV con CVP suprimida la simplificación del TAR a DTG/3TC ha demostrado una eficacia no inferior al cambio a BIC/FTC/TAF, con un incremento de peso tras 48 semanas de tratamiento significativamente menor en aquellos que recibieron DTG/3TC26. Sin embargo, en otro estudio reciente, donde se comparó el cambio a DTG/3TC frente a mantener una pauta con BIC/FTC/TAF, no se observaron diferencias estadísticamente significativas en el cambio de peso entre las dos ramas27. Por ello, los datos actuales no son suficientes para recomendar un cambio de tratamiento antirretroviral ante el aumento de peso corporal. Por el momento, el aumento de peso debe valorarse de forma individualizada y realizarse un abordaje multifactorial28.
4.2.3 Gestión de las toxicidades activas
El cambio de TAR debido a toxicidad es un cambio reactivo, generalmente obligado y, cuyo objetivo es mejorar o resolver dicha toxicidad en la medida en que ésta sea reversible Tabla 1 . Las toxicidades pueden ser sintomáticas y comprometer la adherencia (por ej. los EA del SNC) o pueden ser asintomáticas y, por tanto, no tener impacto en la adherencia (por ej. las toxicidades renal u ósea).
Recomendaciones
- Debe evaluarse de forma activa, en cada visita, la potencial toxicidad del TAR y su impacto en la calidad de vida, incluso si la adherencia es adecuada y la CVP está suprimida (A-III).
- Se debe individualizar el cambio de TAR por toxicidad teniendo en cuenta la gravedad del EA (leve vs grave, subclínico vs clínico), su riesgo de empeoramiento si se mantiene (toxicidad acumulativa vs transitoria), su repercusión en la calidad de vida, las alternativas terapéuticas que permitan mantener la eficacia y la evidencia científica disponible sobre las opciones de cambio (A-III).
- Antes de un cambio de TAR debido a toxicidad o prevención de ésta se debe valorar siempre si los nuevos fármacos o familias pueden compartir efectos secundarios y asegurar que se mantendrá la eficacia virológica (A-III).
4.2.4 Gestión de las interacciones
La mayoría de los ARVs utilizados en la actualidad, tienen un riesgo muy bajo de interacciones medicamentosas. No obstante, éstas pueden constituir un problema en la práctica clínica como consecuencia de la polifarmacia, asociada al envejecimiento e incremento de comorbilidades en la población con VIH, el consumo de drogas recreativas (por ejemplo, chemsex) o el uso de medicamentos de venta libre sin prescripción médica (por ejemplo, suplementos nutricionales o productos naturales).
Recomendaciones
- Se recomienda vigilar activamente las potenciales interacciones farmacológicas con los ARVs y valorar los cambios de TAR proactivos en PVV con comorbilidades y mayor riesgo de interacciones (B-III).
- Siempre que sea posible se recomienda el uso de ARVs no potenciados por su menor riesgo de interacciones (A-III).
- Ante un cambio del TAR, es recomendable comprobar el perfil de interacciones. Para ello existen webs actualizadas de acceso libre (Consultar capítulo 6.5) (A-III).
4.2.5 Deseo gestacional o embarazo en curso
En el capítulo 6.1 se recogen las pautas de TAR recomendadas en la persona con deseo gestacional o embarazo en curso y se revisan los cambios recomendados cuando ya recibe tratamiento. Los principales motivos para realizar un cambio de TAR en este contexto son evitar fármacos o combinaciones con falta de información sobre eficacia y seguridad durante el embarazo o evitar fármacos que pueden presentar niveles plasmáticos insuficientes durante el segundo y tercer trimestre del mismo.
Los cambios proactivos en la PVV con deseo gestacional permiten valorar la tolerancia y eficacia de la nueva pauta de forma anticipada, asegurando la conveniencia del TAR que se mantendrá durante la gestación. En la Tabla 1 se presentan las principales recomendaciones de cambios en este contexto. (Consultar el “Documento de consenso para el seguimiento de la infección por el VIH en relación con la reproducción, embarazo, parto y profilaxis de la transmisión vertical del niño expuesto” en: https://gesida-seimc.org/wp-content/uploads/2024/01/Documento-de-consenso-para-el-seguimiento-de-la-infeccion-por-vih-en-relacion-con-reproducion-embarazo-parto.pdf).
Recomendaciones
- En PVV en TAR embarazadas o con deseo gestacional, debe revisarse el tratamiento para valorar la necesidad de cambiarlo a pautas recomendadas o consideradas más seguras durante el embarazo, y la necesidad de ajustar la dosificación, para garantizar la eficacia virológica de la nueva pauta (A-III).
4.2.6 Cambio de vía de administración y dosificación de acción prolongada
En la actualidad es posible la administración del TAR en una formulación de acción prolongada (CAB+RPV) por vía IM cada 2 meses. Esta nueva estrategia se ha mostrado eficaz como simplificación en pacientes con TAR estable, CVP suprimida (3-6 meses, no se ha establecido un tiempo determinado), sin FV previos a ITINN o INI, sin resistencia documentada o sospechada a RPV o CAB y sin coinfección por el VHB12.
El TAR con CAB+RPV IM puede ofrecer algunas ventajas respecto al TAR por vía oral. Por un lado, permite reducir la frecuencia de dosis, eliminar la toxicidad gastrointestinal o la dificultad para las tomas en personas con problemas de deglución. En el caso de RPV, evita las limitaciones del uso de este fármaco por vía oral (la necesidad de toma con alimentos o la contraindicación del uso de inhibidores de la bomba de protones). Por otro lado, puede favorecer la reducción de la percepción del estigma o la preocupación por la confidencialidad.
Por tanto, la pauta CAB+RPV IM cada 2 meses es una opción adecuada para PVV con CVP suprimida que, de esta forma, puedan mejorar la adherencia, problemas relacionados con la toma del TAR por vía oral (deglución, tolerabilidad digestiva, interacciones, etc.) o aspectos relacionados con la calidad de vida.
El cambio a CAB+RPV cada 2 meses por vía IM puede realizarse directamente desde cualquier pauta de TAR, siendo opcional el periodo de inicio de un mes con CAB y RPV por vía oral29.
Si se plantea un cambio a un TAR con CAB+RPV cada 2 meses IM, hay que tener en cuenta las siguientes consideraciones:
- El principal inconveniente de esta pauta son las reacciones en el lugar de inyección, si bien son habitualmente leves, de corta duración y causan el abandono en menos del 2% de las personas que lo reciben. 1229
- El riesgo de desarrollar MR en caso de FV es muy bajo (1%, habitualmente en el primer año), pero mayor que con pautas orales basadas en otros INI de segunda generación como DTG o BIC, y se han observado algunos casos en personas con adherencia completa al TAR. Análisis a partir de los datos de EC han identificado un mayor riesgo de FV cuando existen MR a RPV archivadas en el ADN proviral, en personas con subtipo de VIH-1 A6 y en aquellas con un IMC ≥30Kg/m2. Se ha estimado que el riesgo de FV en ausencia de estos factores es 0,4% mientras que asciende a 2,0% con un factor y a 19,3% con 2 factores30.
- Los niveles valle bajos (inferior al primer cuartil) de RPV y/o CAB en la semana 8 se han asociado a FV y pueden estar en relación con un IMC >30 Kg/m2 29. En estos casos se recomienda el uso de agujas de inyección de longitud adecuada. Debe explorarse la presencia de prótesis u otras características de la zona glútea que puedan requerir modificar la técnica de inyección.
- Debe garantizarse la adherencia a las visitas para la administración del tratamiento cada 2 meses. En caso de necesidad se puede plantear de forma programada un periodo transitorio (puente) de tratamiento por vía oral.
- En aquellas PVV que cambian a CAB+RPV IM, no hay evidencia de que la monitorización de la CVP deba ser más frecuente que en los cambios a pautas orales en pacientes con CVP suprimida.31
Recomendaciones
- En PVV en las que se considere que la administración IM con formulación de acción prolongada pueda tener beneficios en aspectos clínicos o de calidad de vida y sin contraindicación para el uso de CAB o RPV, el tratamiento con CAB+RPV IM cada 2 meses puede ser una buena opción (A-I).
- No se recomienda el uso de CAB+RPV IM en PVV con evidencia de mutaciones que comprometan la actividad de CAB, RPV o FV con una pauta basada en ITINN o INI (A-I).
- En general, no se recomienda el uso de CAB+RPV IM en PVV con subtipo A6 del VIH-1 (B-I).
- No se considera necesaria la monitorización de niveles plasmáticos de CAB o RPV en la práctica clínica, pero se recomienda adaptar las técnicas y materiales de inyección (tamaño de la aguja) al IMC de las personas tratadas con CAB+RPV IM (A-III).
- Si se interrumpe el TAR con CAB+RPV IM de administración bimestral debe iniciarse una pauta de TAR oral 2 meses después de la última inyección IM (A-I).
- Los controles de CVP tras el cambio a CAB+RPV IM deben realizarse de forma similar a los cambios a pautas de TAR oral (A-I).
Tablas:
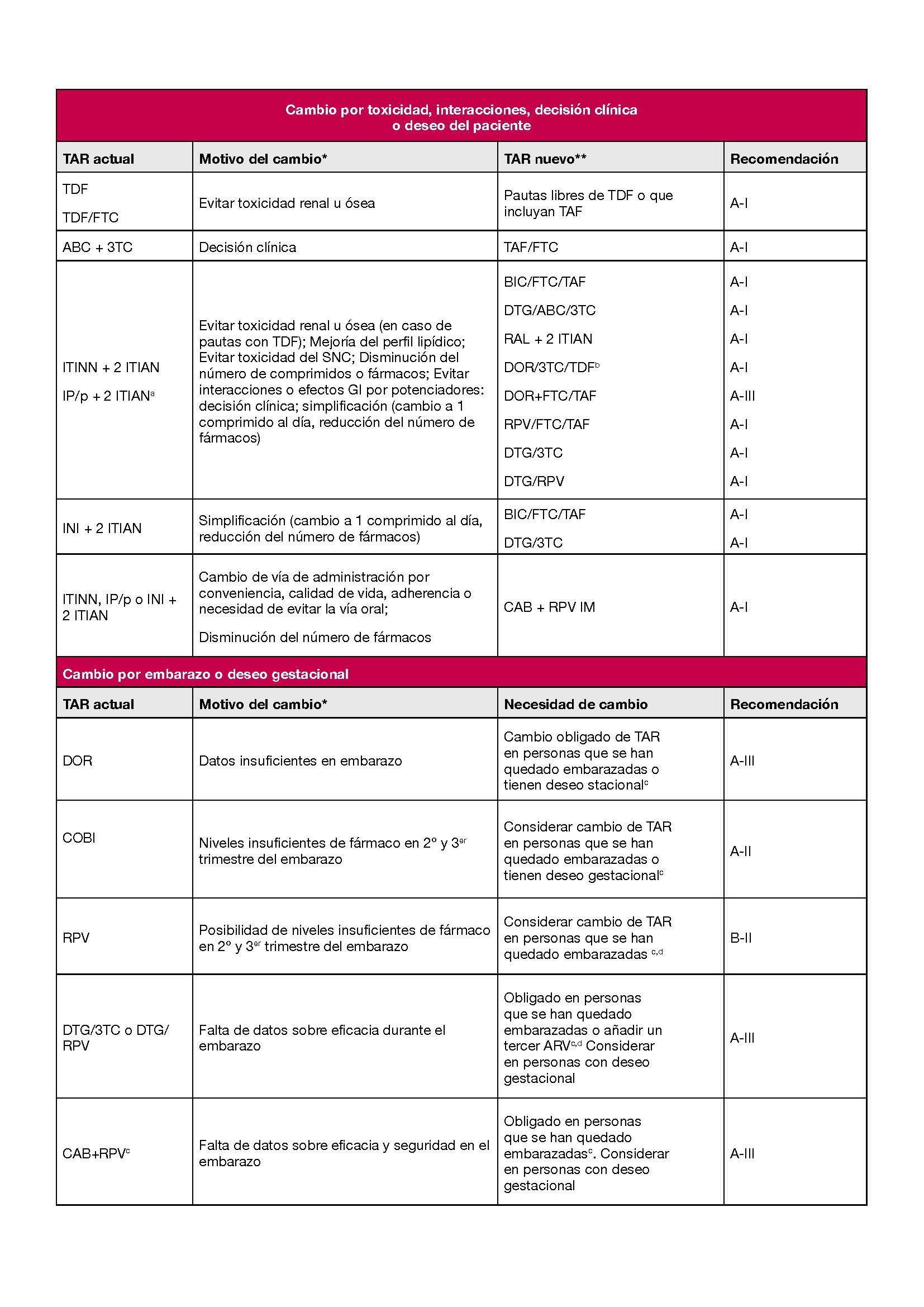
* Véase en el texto la justificación de los motivos del cambio.
** Por orden alfabético dentro de cada familia (INI, ITINN, IP)
a: El cambio desde una pauta con dos ITIAN más un IP/r a dos ITIAN más un ITINN o INI de baja barrera genética solo debe hacerse si se puede garantizar la actividad antiviral de todos los fármacos de la nueva pauta.
b: El comprimido único de DOR/3TC/TDF está aprobado por la AEMPS, pero no está comercializado en España
c: Cambiar a pautas recomendadas durante el embarazo
d: Si se decide mantener, deben realizarse controles frecuentes de CV (cada 1-2 meses) durante el embarazo
Bibliografía:
Fernández A, Imaz A. Clinical considerations when switching antiretroviral therapy. Expert Rev Clin Pharmacol. 2024 Jul;17(7):565-577. https://www.tandfonline.com/doi/full/10.1080/17512433.2024.2365826.
Vasishta S, Dieterich D, Mullen M, et al. Brief Report: Hepatitis B Infection or Reactivation After Switch to 2-Drug Antiretroviral Therapy: A Case Series, Literature Review, and Management Discussion. J Acquir Immune Defic Syndr. 2023 Oct;94(2):160-164. https://journals.lww.com/jaids/abstract/2023/10010/brief_report__hepatitis_b_infection_or.9.aspx.
Molina JM, Ward D, Brar I, et al. Switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from dolutegravir plus abacavir and lamivudine in virologically suppressed adults with HIV-1: 48 week results of a randomised, double-blind, multicentre, active-controlled, phase 3, non-inferiority trial. Lancet HIV. 2018 Jul;5(7):e357-e365. https://www.thelancet.com/journals/lanhiv/article/PIIS2352-3018(18)30092-4/abstract.
Daar ES, DeJesus E, Ruane P et al. Efficacy and safety of switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from boosted protease inhibitor-based regimens in virologically suppressed adults with HIV-1: 48-week results of a randomised, open-label, multicenter, phase 3, non-inferiority trial. Lancet HIV 2018;5: e347-e356. https://www.thelancet.com/journals/lanhiv/article/PIIS2352-3018(18)30091-2/fulltext
Hagins D, Kumar P, Saag M, et al; BRAAVE2020 Investigators. Switching to Bictegravir/Emtricitabine/Tenofovir Alafenamide in Black Americans with HIV-1: A Randomized Phase 3b, Multicenter, Open-Label Study. J Acquir Immune Defic Syndr. 2021 Sep 1;88(1):86-95. https://journals.lww.com/jaids/fulltext/2021/09010/switching_to_bictegravir_emtricitabine_tenofovir.12.aspx.
Osiyemi O, De Wit S, Ajana F, et al. Efficacy and safety of switching to Dolutegravir/Lamivudine (DTG/3TC) versus continuing a Tenofovir Alafenamide-based 3- or 4-drug regimen for maintenance of virologic suppression in adults living with HIV-1: results through week 144 from the Phase 3, Non-inferiority TANGO Randomized Trial. Clin Infect Dis 2022; 75: 975-86.
Llibre JM, Brites C, Cheng CY, et al. Efficacy and Safety of Switching to the 2-Drug Regimen Dolutegravir/Lamivudine Versus Continuing a 3- or 4-Drug Regimen for Maintaining Virologic Suppression in Adults Living With Human Immunodeficiency Virus 1 (HIV-1): Week 48 Results from the Phase 3, Noninferiority SALSA Randomized Trial. Clin Infect Dis. 2023 Feb 18;76(4):720-729. https://academic.oup.com/cid/article/76/4/720/6541246?login=false"%20\t%20"_blank.
Aboud M, Orkin C, Podzamczer D, et al. Efficacy and safety of dolutegravir-rilpivirine for maintenance of virological suppression in adults with HIV-1: 100-week data from the randomised, open-label, phase 3 SWORD-1 and SWORD-2 studies. Lancet HIV 2019; 6: e576–e587.
Palella FJ Jr, Fisher M, Tebas P, et al. Simplification to rilpivirine/emtricitabine/tenofovir disoproxil fumarate from ritonavir-boosted protease inhibitor antiretroviral therapy in a randomized trial of HIV-1 RNA-suppressed participants. AIDS 2014; 28:335-44.
Johnson M, Kumar P, Molina JM, et al. Switching to Doravirine/Lamivudine/Tenofovir Disoproxil Fumarate (DOR/3TC/TDF) maintains HIV-1 virologic suppression through 48 Weeks: Results of the DRIVE-SHIFT Trial. J Acquir Immune Defc Syndr 2019; 81:463-72.
Eron JJ, Orkin C, Cunningham D, et al; EMERALD study group. Week 96 efficacy and safety results of the phase 3, randomized EMERALD trial to evaluate switching from boosted-protease inhibitors plus emtricitabine/tenofovir disoproxil fumarate regimens to the once daily, single-tablet regimen of darunavir/cobicistat/emtricitabine/ tenofovir alafenamide (D/C/F/TAF) in treatment-experienced, virologically suppressed adults living with HIV-1. Antiviral Res 2019;170. https://pubmed.ncbi.nlm.nih.gov/31279073/
Overton ET, Richmond G, Rizzardini G, et al. Long-acting cabotegravir and rilpivirine dosed every 2 months in adults with HIV-1 infection (ATLAS-2M), 48-week results: randomised, multicenter, open-label, phase 3b, non-inferiority study. Lancet 2021; 396: 1994-2005.
Sax PE, Andreatta K, Molina JM, et al. High efficacy of switching to bictegravir/emtricitabine/tenofovir alafenamide in people with suppressed HIV and preexisting M184V/I. AIDS. 2022 Sep 1;36(11):1511-1520.
Sax PE, Rockstroh JK, Luetkemeyer AF, et al. Switching to Bictegravir, Emtricitabine, and Tenofovir Alafenamide in Virologically Suppressed Adults With Human Immunodeficiency Virus. Clin Infect Dis. 2021 Jul 15;73(2):e485–e493. https://academic.oup.com/cid/article/73/2/e485/5872016?login=false" \t "_blank.
Jary A, Marcelin AG, Charpentier C, et al. M184V/I does not impact the efficacy of abacavir/lamivudine/dolutegravir use as switch therapy in virologically suppressed patients. J Antimicrob Chemother 2020; 75:1290-3.
Ombajo LA, Penner J, Nkuranga J, et al. Second-Line Switch to Dolutegravir for Treatment of HIV Infection. N Engl J Med. 2023 Jun 22;388(25):2349-2359. https://www.nejm.org/doi/10.1056/NEJMoa2210005?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed.
De Miguel R, , Sigcha M, de Lagarde M, et al. Impact of Archived Minority Populations With M184V/I on DTG/3TC for Maintenance of Viral Suppression. [poster]. Conference on Retroviruses and Opportunistic Infections. March 3-6, 2024, Denver, Colorado. #693. https://www.croiconference.org/wp-content/uploads/sites/2/posters/2024/693.pdf
Podzamczer D, Imaz A, Lopez-Lirola A, et al. Switching to bictegravir/emtricitabine/tenofovir alafenamide (BIC/FTC/TAF) plus darunavir/cobicistat in heavily antiretroviral-experienced, virologically suppressed HIV-infected adults receiving complex regimens. J Antimicrob Chemother 2023; 78:2696-2701.
Spinner CD, Kümmerle T, Schneider J, et al. Efficacy and safety of switching to dolutegravir with boosted darunavir in virologically suppressed adults with HIV-1: A Randomized, Open-Label, Multicenter, Phase 3, Noninferiority Trial: The DUALIS Study. Open Forum Infect Dis 2020; 7:1-8.
Raffi F, Orkin C, Clarke A, et al. Brief Report: Long-Term (96-Week) Efficacy and Safety After Switching From Tenofovir Disoproxil Fumarate to Tenofovir Alafenamide in HIV-Infected, Virologically Suppressed Adults. J Acquir Immune Defic Syndr. 2017 Jun 1;75(2):226-231. https://journals.lww.com/jaids/fulltext/2017/06010/brief_report__long_term__96_week__efficacy_and.12.aspx.
Casado JL, Santiuste C, Vivancos MJ, et al. Switching to abacavir versus use of a nucleoside-sparing dual regimen for HIV-infected patients with tenofovir-associated renal toxicity. HIV Med 2018;541-550.
Tebas P, Sension M, Arribas JR, et al; ECHO and THRIVE Study Groups. Lipid Levels and Changes in Body Fat Distribution in Treatment-Naive, HIV-1-Infected Adults Treated with Rilpivirine or Efavirenz for 96 Weeks in the ECHO and THRIVE Trials. Clin Infect Dis. 2014 Aug 1;59(3):425-434. https://academic.oup.com/cid/article/59/3/425/2895310?login=false.
Gatell JM, Assoumou L, Moyle G, et al. Immediate vs. deferred switching from a boosted protease inhibitor-based regimen to a dolutegravir based regimen in virologically suppressed patients with high cardiovascular risk or age >50 years: Final 96 weeks results of NEAT 022 study. Clin Infect Dis 2019; 68:597-606.
Martinez E, Larrousse M, Llibre JM, et al. Substitution of raltegravir for ritonavir-boosted protease inhibitors in HIV-infected patients: the SPIRAL study. AIDS 2010; 24: 1697-1707.
van Wyk J, Ait-Khaled M, Santos J, et al. Improvement in metabolic health parameters at Week 48 after switching from a tenofovir alafenamide-based 3- or 4-drug regimen to the 2-drug regimen of dolutegravir/lamivudine: The TANGO Study. J Acquir Immune Defic Syndr 2021; 87:794-800.
Ryan P, Blanco JL, Masiá M, et al. Non-inferior efficacy and less weight gain when switching to DTG/3TC than when switching to BIC/FTC/TAF in virologically suppressed people with HIV (PWH): the PASO-DOBLE (GeSIDA 11720) randomised clinical trial. [poster]. 25th International AIDS Conference, AIDS 2024. Munich, 22-26 July 2024. #12253. https://programme.aids2024.org/Abstract/Abstract/?abstractid=12253.
Rolle CP, Castano J, Nguyen V, et al. Efficacy, Safety, and Tolerability of Switching From Bictegravir/Emtricitabine/Tenofovir Alafenamide to Dolutegravir/Lamivudine Among Adults With Virologically Suppressed HIV: The DYAD Study. Open Forum Infect Dis. 2024 Sep 26;11(10):ofae560. https://academic.oup.com/ofid/article/11/10/ofae560/7777137?login=false.
Wohl DA, Koethe JR, Sax PE, et al. Antiretrovirals and Weight Change: Weighing the Evidence. Clin Infect Dis. 2024 Oct 15;79(4):999-1005. https://academic.oup.com/cid/article-abstract/79/4/999/7644593?redirectedFrom=fulltext&login=false.
Orkin C, Bernal Morell E, Tan DHS, et al. Initiation of long-acting cabotegravir plus rilpivirine as direct-to-injection or with an oral lead-in in adults with HIV-1 infection: week 124 results of the open-label phase 3 FLAIR study. Lancet HIV. 2021 Nov;8(11):e668-e678. https://www.sciencedirect.com/science/article/abs/pii/S2352301821001843?via%3Dihub.
Orkin C, Schapiro JM, Perno CF, et al. Expanded Multivariable Models to Assist Patient Selection for Long-Acting Cabotegravir + Rilpivirine Treatment: Clinical Utility of a Combination of Patient, Drug Concentration, and Viral Factors Associated With Virologic Failure. Clin Infect Dis. 2023 Nov 17;77(10):1423-1431. https://academic.oup.com/cid/article/77/10/1423/7204256?login=true.
Kito C, Mambule IK, Musaazi J, et al; CARES trial team. Switch to long-acting cabotegravir and rilpivirine in virologically suppressed adults with HIV in Africa (CARES): week 48 results from a randomised, multicentre, open-label, non-inferiority trial. Lancet Infect Dis. 2024 Oct:24(10):1083-1092. doi: 10.1016/S1473-3099(24)00289-5. https://www.sciencedirect.com/science/article/abs/pii/S1473309924002895?via%3Dihub.
5. FRACASO DEL TRATAMIENTO ANTIRRETROVIRAL
5.1. DEFINICIONES
5.2. CONSIDERACIONES GENERALES
5.3. ESCENARIOS CLÍNICOS DE FRACASO VIROLÓGICO
DEFINICIONES
- Fracaso virológico (FV): CVP >200 cop/mL transcurridas 24 semanas desde el inicio del TAR, confirmada en una segunda muestra consecutiva.
- Repuntes virológicos transitorios aislados (“blips”): CVP entre 50-200 cop/mL, con valores de CVP previa y posterior <50 cop/mL. Los “blips” aislados no parecen tener repercusión clínica1.
- Viremia de bajo nivel (VBN): CVP 50-200 cop/mL en al menos dos muestras consecutivas. En estos casos hay que revisar la adherencia al tratamiento, así como potenciales interacciones con otros tratamientos concomitantes o productos farmacológicos. Si ya el paciente realiza un TAR ajustado al historial de resistencias de los estudios genotípicos disponibles, otras estrategias terapéuticas como el cambio o intensificación del TAR no han demostrado beneficios.
CONSIDERACIONES GENERALES
Las tasas de incidencia de FV de los TAR de inicio preferentes a las 48 semanas son inferiores al 5%. Los factores que influyen en el FV son:
- Mala adherencia al tratamiento o al seguimiento de controles médicos.
- Efectos adversos (EA).
- Interacciones farmacocinéticas (fármacos, productos de herboristería, alimentos, complementos nutricionales o drogas recreativas).
- Preexistencia de MR.
El objetivo del TAR de rescate es volver a suprimir la CVP (<50 cop/mL) tan pronto como sea posible. Volver a suprimir la CVP consigue los siguientes objetivos adicionales:
- Evitar la acumulación de MR, la resistencia cruzada y la evolución viral.
- Reconstituir el sistema inmunitario.
- Disminuir la inflamación crónica.
- Disminuir eventos definitorios y no definitorios de SIDA.
- Reducir la mortalidad.
- Reducir el riesgo de transmisión del VIH.
Recomendaciones
- El cambio del TAR por FV debe efectuarse precozmente para evitar la acumulación de MR y facilitar la respuesta al nuevo tratamiento (A-III).
- Se deben analizar las causas que motivaron el fracaso, la historia farmacológica y los fracasos previos. El nuevo TAR debe ser lo más cómodo y bien tolerado posible (A-III).
- El nuevo TAR debe contener al menos 2 fármacos completamente activos y, al menos uno de ellos, debe tener una alta barrera a las resistencias (DRV/p o INI de segunda generación). Añadir un cuarto fármaco no aporta beneficios, especialmente en PVV en las que no se detectan MR (A-II).
- En los pacientes que necesitan tratamiento para VHB en que se modifique el TAR por FV debemos evitar rebrotes de replicación, asegurando que el tratamiento de la infección por VHB no se discontinúa (A-I).
- En sujetos con VBN, tras revisar adherencia, potenciales interacciones y TAR ajustado al historial de resistencias, se recomienda mantener la pauta con una vigilancia estrecha (B-III)
ESCENARIOS CLÍNICOS DE FRACASO VIROLÓGICO
5.3.1. Fracaso virológico precoz
Es el FV a la primera línea de TAR.
Recomendaciones
- Se adecuarán los cambios de TAR a las MR para conseguir un TAR de rescate plenamente activo (A-II).
- Además de investigar MR, se deberá descartar mala adherencia y se investigarán posibles interacciones, errores en la prescripción o la toma, y otros motivos en la vida del paciente que dificulten una correcta toma de la medicación (A-II).
- En ausencia de MR, y una vez corregido lo expuesto arriba, se podrá continuar el mismo TAR con un control precoz de la CVP (1-3 meses dependiendo de la CVP) (C-III).
- Alternativamente- especialmente en FV a esquemas con baja barrera a las resistencias- se modificará el TAR según lo resumido en la Tabla 1 (B-I).
5.3.2. Fracaso virológico avanzado
Es el FV a la segunda o sucesivas líneas de TAR. En este escenario, la mayoría de los pacientes pueden presentan MR a dos o más familias de FAR.
Los principales EC de pautas de rescate avanzado se resumen en la Tabla 1 223.
Recomendaciones
- Para el diseño de un tratamiento de rescate se tendrán en cuenta todos los genotipos disponibles (genotipo acumulado), las pautas previas y las MR no recogidas en los estudios previos que pudieran estar presentes (A-I).
- Todo TAR de rescate avanzado incluirá DRV/p y/o DTG, si persiste susceptibilidad viral a estos fármacos (A-I).
- Cuando existe alguna mutación mayor de resistencia a DRV, se recomienda utilizar DRV/r 600/100 mg BID. DRV/p deberá acompañarse de otros FAR (ITIAN, ITINN e INI) hasta conseguir un régimen plenamente activo (A-I).
- El INI de elección para TAR de rescate es DTG BID (QD únicamente si no existe ninguna MR en la integrasa ni exposición previa a INI) (A-I). Bictegravir coformulado con TAF/FTC puede ser una alternativa en PPV en las que no se detecten MR en la integrasa ni exposición previa a INI (B-III).
- En general se recomienda mantener XTC incluso en presencia de M184V (B-III).
- Si no existen MR que reduzcan su eficacia, se puede considerar añadir un ITINN de segunda generación al régimen de rescate, como ETV (B-I) o DOR (B-III).
- En caso de que, con los ITIAN, ITINN, IP e INI, no se pueda construir un esquema con garantías de supresión virológica completa y sostenida se deberá incluir en el tratamiento de rescate un fármaco que actúe en otras dianas virales (inhibidor de fusión, inhibidor del acoplamiento, inhibidor de la cápside o inhibidor de la translocación de la transcriptasa inversa) (A-I).
- Si el régimen de rescate final es complejo, incluye múltiples fármacos y existe riesgo de que la adherencia a largo plazo se comprometa, es posible plantear la reducción de dicha complejidad, retirando algún fármaco o pasando a una pauta QD (C-III). Esta decisión no está exenta de riesgo por lo que se deberá realizar por un médico experto en el manejo de pacientes complejos, valorando cuidadosamente el beneficio/riesgo y únicamente tras conseguir la supresión virológica completa durante un mínimo de 6 meses (A-III).
5.3.3. Fracaso virológico en pacientes con escasas opciones terapéuticas
Existen datos de nuevos fármacos que actúan en diferentes dianas del virus:
- Fostemsavir (FTV): inhibidor del acoplamiento (attachment) y prodroga del Temsavir que se administra a dosis de 1 comprimido de 600 mg BID. Ha demostrado en el estudio BRIGHTE22 una tasa de respuesta (CVP <40 cop/mL) en semana 96 del 60% en la cohorte randomizada (CR: posibilidad de usar al menos 1 fármaco activo aprobado) y del 37% en la cohorte no randomizada (NR: sin posibilidad de usar fármacos activos aprobados). Con una mediana basal de 80 linfocitos T CD4+, a la semana 96 la recuperación media fue de 205 células/μL en la CR y de 119 células/μL en la NR. Aprobado por la FDA y la EMA.
- Lenacapavir (LEN): inhibidor de la cápside administrado por vía subcutánea cada 6 meses. El estudio CAPELLA24 demostró excelente actividad antiviral de LEN combinado con un régimen basal optimizado (TO) en 72 pacientes extensamente pretratados y virus multirresistente, presentando supresión virológica en semana 156 el 85% de los pacientes. Aprobado por la FDA y la EMA.
Recomendaciones
- No interrumpir el TAR (A-I).
- Se recomienda derivar al paciente a un centro con experiencia y acceso a nuevos FAR a través de ensayos o programas de acceso expandido, que puedan estar disponibles (A-III).
- Se debe evitar exponer al paciente a monoterapia funcional con un solo FAR por el elevado riesgo de FV y selección de MR a ese único FAR (A-III).
- En el caso excepcional de que se agoten todas las opciones terapéuticas, se recomendará un tratamiento “puente” que combine efectividad residual de los fármacos, permita una supresión parcial de la replicación viral, y en lo posible, minimice la acumulación de MR que limiten la efectividad de futuros fármacos (A-III).
- En cuanto sea posible, este tratamiento debe cambiarse a un TAR supresor con 2-3 FAR activos que incluya fármacos de nuevas familias. (A-III)
Tablas:
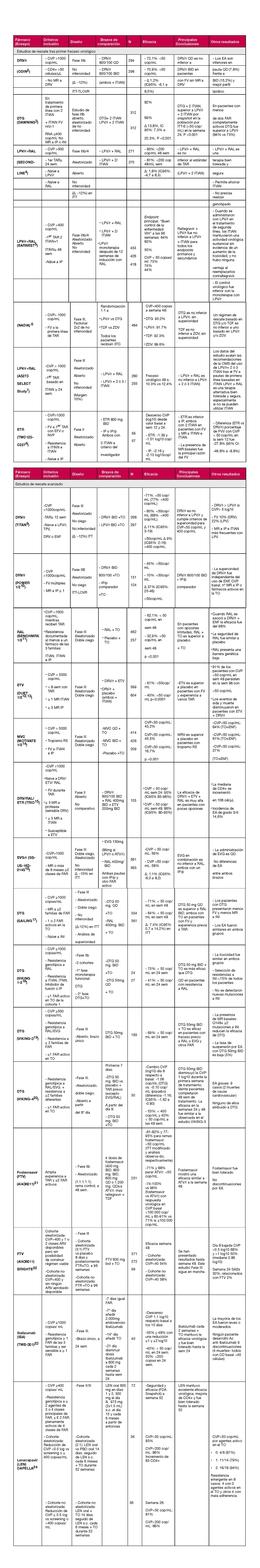
Bibliografía:
Saag MS. What's All This Fuss I Hear About Viral "Blips"? Clin Infect Dis. 2020 Jun 10;70(12):2710-2711.
Sension M, Cahn P, Domingo P, et al. Subgroup analysis of virological response rates with once- and twice-daily darunavir/ritonavir in treatment-experienced patients without darunavir resistance-associated mutations in the ODIN trial. HIV Med 2013; 14:437-44.
Aboud M, Kaplan R, Lombaard J, et al. Dolutegravir versus ritonavir-boosted lopinavir both with dual nucleoside reverse transcriptase inhibitor therapy in adults with HIV-1 infection in whom first-line therapy has failed (DAWNING): an open-label, non-inferiority, phase 3b trial. Lancet Infect Dis 2019; 19:253-64.
Boyd MA, Kumarasamy N, Moore CL, et al. Ritonavir-boosted lopinavir plus nucleoside or nucleotide reverse transcriptase inhibitors versus ritonavir-boosted lopinavir plus raltegravir for treatment of HIV-1 infection in adults with virological failure of a standard first-line bART regimen (SECOND-LINE): a randomised, open-label, non-inferiority study. Lancet 2013; 381:2091-9.
Paton N, Kityo C, Thompson J, et al. Nucleoside reverse-transcriptase inhibitor cross-resistance and outcomes from second-line antiretroviral therapy in the public health approach: an observational analysis within the randomised, open-label, EARNEST trial. Lancet HIV 2017;4: e341-e348.
Paton NI, Musaazi J, Kityo C, et al. Efficacy and safety of dolutegravir or darunavir in combination with lamivudine plus either zidovudine or tenofovir for second-line treatment of HIV infection (NADIA): week 96 results from a prospective, multicentre, open-label, factorial, randomised, non-inferiority trial. Lancet HIV 2022;9: e381-e393.
La Rosa AM, Harrison LJ, Taiwo B, et al. Raltegravir in second-line antiretroviral therapy in resource-limited settings (SELECT): a randomised, phase 3, non-inferiority study. Lancet HIV 2016; 3: e247-58.
Ruxrungtham K, Pedro RJ, Latiff GH, et al. Impact of reverse transcriptase resistance on the efficacy of TMC125 (etravirine) with two nucleoside reverse transcriptase inhibitors in protease inhibitor-naïve, nonnucleoside reverse transcriptase inhibitor-experienced patients: study TMC125-C227. HIV Med 2008; 9:883-96.
Madruga JV, Berger D, McMurchie M, et al. Efficacy and safety of darunavir-ritonavir compared with that of lopinavir-ritonavir at 48 weeks in treatment-experienced, HIV- infected patients in TITAN: a randomised controlled phase III trial. Lancet 2007; 370:49-58.
Clotet B, Bellos N, Molina JM, et al. Efficacy and safety of darunavir-ritonavir at week 48 in treatment-experienced patients with HIV-1 infection in POWER 1 and 2: a pooled subgroup analysis of data from two randomised trials. Lancet 2007; 369:1169-78.
Steigbigel RT, Cooper DA, Teppler H, et al. Long-term efficacy and safety of raltegravir combined with optimized background therapy in treatment-experienced patients with drug- resistant HIV infection: week 96 results of the BENCHMRK 1 and 2 Phase III trials. Clin Infect Dis 2010; 50:605-12.
Madruga JV, Cahn P, Grinsztejn B, et al. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-1: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet 2007; 370:29-38.
Lazzarin A, Campbell T, Clotet B, et al. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-2: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet 2007; 370:39-48.
Gulick RM, Lalezari J, Goodrich J, et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med 2008; 359:1429-41.
Yazdanpanah Y, Fagard C, Descamps D, et al. High rate of virologic suppression with raltegravir plus etravirine and darunavir/ritonavir among treatment-experienced patients infected with multidrug-resistant HIV: results of the ANRS 139 TRIO trial. Clin Infect Dis 2009; 49:1441-9.
Molina JM, Lamarca A, Andrade-Villanueva J, et al. Efficacy and safety of once daily elvitegravir versus twice daily raltegravir in treatment-experienced patients with HIV-1 receiving a ritonavir-boosted protease inhibitor: randomised, double-blind, phase 3, non-inferiority study. Lancet Infect Dis 2012; 12:27-35.
Cahn P, Pozniak AL, Mingrone H, et al. Dolutegravir versus raltegravir in antiretroviral- experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet 2013; 382:700-8.
Eron JJ, Clotet B, Durant J, et al. Safety and efficacy of dolutegravir in treatment- experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study. J Infect Dis 2013; 207:740-8.
Castagna A, Maggiolo F, Penco G, et al. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the Phase III VIKING-3 Study. J Infect Dis 2014; 210:354-62.
Akil B, Blick G, Hagins DP, et al. Dolutegravir versus placebo in subjects harbouring HIV-1 with integrase inhibitor resistance associated substitutions: 48-week results from VIKING-4, a randomized study. Antivir Ther 2015; 20:343-8.
Thompson M, Lalezari J, Richard R, et.al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug fostemsavir in antiretroviral-experienced subjects: week 48 analysis of AI438011, a Phase IIb, randomized controlled trial. Antivir Ther2017; 22:215-23.
Kozal, J. Aberg, G Pialloux et al: Fostemsavir in Adults with Multidrug-Resistant HIV-1 Infection (BRIGHTE Study). N Engl J Med 2020; 382:1232-43.
Emu B, Fessel J, Schrader S, et al. Phase 3 Study of Ibalizumab for Multidrug-Resistant HIV-1. N Engl J Med 2018; 379:645-54.
Ogbuagu O, McGowan JP, Stapleton A, et al. CAPELLA: Wk 156 Results With LA SC Lenacapavir in Persons Living With Multidrug-Resistant HIV. IDWeek 2024, Oral 155.
6. SITUACIONES CON CONSIDERACIONES ESPECÍFICAS DEL TAR
6.1. EMBARAZO
6.2. TUBERCULOSIS
6.3. HEPATOPATÍAS
6.4. INSUFICIENCIA RENAL
6.5. INTERACCIONES MÁS RELEVANTES Y RECURSOS DISPONIBLES
6.6. INFECCIÓN POR EL VIH-2.
EMBARAZO
Se recomienda la lectura de guías actualizadas1 y el documento de consenso de GeSIDA y el PNS con otras sociedades2.
El objetivo del TAR durante el embarazo es conseguir y mantener CVP indetectable durante el mayor tiempo posible, especialmente en el tercer trimestre y en el momento del parto2. Se recogen las recomendaciones sobre el uso de FAR en el embarazo en la Tabla 1.
Recomendaciones
- La elección de los FAR concretos en las mujeres con deseo gestacional o gestantes se debe basar en la eficacia, seguridad y farmacocinética durante el embarazo. De forma individualizada se tendrá en cuenta el estudio de resistencias y la tolerancia en cada caso. El TAR de elección consiste en la combinación de dos ITIAN: TDF/FTC o TAF/FTC, junto con DTG una vez al día (A-I) o RAL dos veces al día (A-II). Podrán recibir cualquiera de los FAR “recomendados” o “alternativos” tras una valoración individualizada (A-III).
- En mujeres con TAR y CVP indetectable se mantendrá el mismo TAR siempre que haya demostrado seguridad y niveles farmacocinéticos adecuados en embarazadas (A-II).
- En caso de que una mujer esté recibiendo biterapia y se quede embarazada, en general se recomienda el cambio a triple terapia (B-III).
- El tratamiento intraparto con ZDV vía intravenosa está indicado, independientemente del TAR que haya recibido durante el embarazo, si la CVP en el parto es >1000 copias/mL o desconocida (A-I). Se debe considerar si CVP en el parto está entre 50 y 999 copias/mL y si hay dudas de la adherencia al tratamiento (B-III).
TUBERCULOSIS
Se recomienda consultar el Documento de Prevención y Tratamiento de Infecciones Oportunistas y otras Coinfecciones en Pacientes con Infección por VIH (febrero 2022)3.
Recomendaciones de TAR
- a) Inicio
- Iniciar TAR independientemente de la cifra de CD4+ en las dos primeras semanas del inicio del tratamiento antituberculoso, una vez comprobada la tolerancia al mismo y excluyendo la meningitis tuberculosa (A-I).
- En el caso de meningitis tuberculosa, demorar el inicio del TAR al menos 4 semanas, eligiendo el momento óptimo según la situación clínica del paciente (A-I).
- b) Fármacos
- ITIAN. Se pueden utilizar TDF, 3TC, FTC o ABC (A-I). Evitar el uso de TAF con rifampicina (A-II); si fuese la única opción, monitorizar CVP y efectos adversos porque no hay resultados clínicos y hay estudios que observan una mayor concentración intracelular de TFV con esta asociación (C-III).
- Tercer fármaco:
- Según tolerancia: DTG 50 mg/12 h (A-II)6, RAL 800 mg/12 h (A-I) o EFV a dosis estándar (A-I).
- Si excepcionalmente la única opción fuese un IP, sustituir rifampicina por rifabutina con ajuste de dosis (A-I).
- El uso de rifapentina (no comercializada en España) con EFV permitiría administrar RP/Mx/H/ZDV 4 meses en pacientes con CD4>100 células/µl.
- Fármacos que no pueden utilizarse con rifampicina: RPV, ETR, DOR, ningún IP (potenciado o no), COBI, EVG, RAL 1200 mg QD, BIC QD (A-I)5, CAB inyectable, FTV ni LEN6.
Síndrome inflamatorio por reconstitución inmunológica (SIRI)
- No interrumpir el tratamiento antituberculoso ni el TAR (A-III).
- En pacientes con TB y recuento de linfocitos CD4+< 100 células/μL administrar prednisona (40 mg/día 2 semanas y luego 20 mg/día 2 semanas más) en el primer mes tras antituberculosos para prevenir el SIRI (A-I).
- Una vez desarrollado SIRI, para el manejo de los síntomas pueden añadirse AINE en las formas leves o moderadas (A-III) o corticosteroides en las formas graves (A-II).
HEPATOPATÍAS
Se recomienda consultar el documento de consenso específico realizado por GESIDA y GEHEP7.
En PVV con hepatitis crónica por VHB, el estudio ALLIANCE demostró mayor supresión virológica del VHB a las 48 semanas (63% vs 43%) en pacientes que recibían BIC/FTC/TAF frente DTG+TDF/FTC8. Sin embargo, estas tasas tendieron a igualarse en la semana 96. La relevancia clínica de este dato se desconoce.
La Tabla 2 recoge las consideraciones sobre uso de ARV en pacientes con cirrosis hepática79.
Recomendaciones
- En pacientes coinfectados por VHB y/o VHC o con enfermedad hepática metabólica se recomienda iniciar el TAR tan pronto como sea posible (A-I).
- En PVV naïve que precisan tratamiento de la hepatitis C, en general es preferible iniciar primero el TAR (B-III).
- En pacientes coinfectados por VIH/VHB se debe iniciar precozmente un TAR que incluya TAF/FTC como pauta preferente y TDF/FTC como alternativa (A-I).
- En pacientes coinfectados por VIH/VHB se debe evitar la interrupción de una pauta que incluya TAF o TDF (A-II).
- En pacientes con enfermedad hepática metabólica se recomienda usar FAR con perfil lipídico favorable (A-II)10.
- En pacientes con hepatopatía crónica y función hepática conservada, incluida la cirrosis estadio A de Child-Pugh se puede utilizar cualquier FAR (A-I).
- En pacientes con insuficiencia hepatocelular leve/moderada (Child-Pugh A o B), los ITIAN (salvo ABC que no debe usarse en Child-Pugh B), los INI, RPV, DOR o DRV no precisan ajuste de dosis y son los fármacos de elección (A-II).
- En pacientes en estadio de Child-Pugh C, están contraindicados el ABC y los IP potenciados (A-II) y deben considerarse preferentes las pautas basadas en RAL (AII), DTG o BIC (BIII).
INSUFICIENCIA RENAL
Se recomienda consultar el documento de consenso elaborado por GeSIDA11, así como otras citas recientes. La dosificación de los fármacos según el filtrado glomerular estimado (FGe) se expone en la Tabla 3.
Recomendaciones
- Es necesario ajustar las dosis de los ITIAN, excepto en el caso de ABC. (A-II).
- Hay que ajustar la dosis de MVC si se emplea en combinación con inhibidores potentes del CYP3A4. (A-II)
- No se requiere ajuste de dosis de los ITINN12 (en pacientes con insuficiencia renal avanzada en tratamiento con RPV se recomienda vigilancia estrecha por el potencial aumento de sus concentraciones plasmáticas), los IP, los INI RAL, DTG o CAB, los inhibidores de la entrada, ibalizumab y fostemsavir, ni del inhibidor de la cápside lenacapavir13, incluso en pacientes con insuficiencia renal severa o en hemodiálisis. En el caso de DTG se debe tener precaución con la coadministración de cationes. (A-II)
- En general, se desaconseja el uso de coformulaciones de FAR en los que alguno de los fármacos precise ajuste de dosis en pacientes con insuficiencia renal significativa incluyendo pacientes en hemodiálisis Tabla 3. En estos casos, deben emplearse los FAR por separado y realizar los ajustes pertinentes. (B-II)
- En los pacientes en TAR con cualquier grado de insuficiencia renal se recomienda evitar los fármacos nefrotóxicos y realizar una vigilancia estrecha de la función renal. (A-III)
INTERACCIONES MÁS RELEVANTES Y RECURSOS DISPONIBLES
Recomendaciones
- Se debe elaborar una historia farmacológica completa que incluya el tratamiento antirretroviral, co-medicaciones prescritas por otros médicos u otras sustancias que el paciente pueda tomar sin receta médica, tratamientos complementarios (suplementos, anabolizantes, fitoterapia, etc) y/o drogas recreativas. Esta historia farmacológica debe actualizarse periódicamente (A-III).
- Se debe pensar en la posibilidad de una interacción medicamentosa ante cualquier modificación del plan terapéutico, tanto antes de iniciar un nuevo medicamento como al interrumpir un fármaco ya en uso (A-III).
- Ante la posibilidad de una interacción medicamentosa, se deben consultar recursos específicos. Las fichas técnicas de los medicamentos también son de gran utilidad (A-III).
- Web de la Universidad de Liverpool: www.hiv-druginteractions.org
- Up to Date. Posibilidad de consultar interacciones con el tratamiento antirretroviral y entre otros medicamentos (requiere suscripción): https://www.wolterskluwer.com/es-es/solutions/uptodate
- Casos clínicos reales sobre interacciones en la práctica clínica: www.clinicalcasesDDIs.com
- Hospital General de Toronto. Contiene información extensa sobre interacciones con quimioterápicos. https://hivclinic.ca/drug-information/antiretroviral-interactions-with-chemotherapy-regimens/
- Kaiser Permanente. Contiene información extensa sobre interacciones con fitoterapia y otros tratamientos alternativos. https://healthy.kaiserpermanente.org/health-wellness/natural-medicines
- Por su menor riesgo de causar interacciones, se debe priorizar el uso de esquemas antirretrovirales basados en INI no potenciados (BIC, DTG, RAL). DOR también puede ser de utilidad en este escenario (A-I).
- La administración IM de CAB y RPV presenta la ventaja de evitar interacciones a nivel del tubo digestivo (inhibidores de la bomba de protones con RPV, cationes divalentes con los INI, inhibición o inducción de enzimas y transportadores de fármacos a nivel intestinal). Sin embargo, su administración conjunta con inductores potentes de CYP3A4 y/o UGT (rifampicina, antiepilépticos, etc.) conlleva a una reducción significativa de la exposición a CAB y a RPV, lo que puede conllevar un aumento del riesgo de FV.
- Cuando se usen fármacos inductores (rifampicina, antiepilépticos…) o inhibidores (antifúngicos, ciclosporina…) de los sistemas enzimáticos o de los transportadores involucrados en la farmacocinética de los antirretrovirales, se debe revisar cuidadosamente el esquema antirretroviral. Se debe considerar la magnitud de la interacción, el intervalo terapéutico, la duración de la coadministración y las alternativas de tratamiento válidas en cada paciente (A-I).
- Para el manejo de aquellos pacientes complejos que reciben múltiples fármacos con posibles interacciones y/o con un intervalo terapéutico estrecho, como es el caso de los pacientes oncológicos, se recomienda un abordaje multidisciplinar que involucre a especialistas de cada área terapéutica, en el tratamiento del VIH y a farmacéuticos/farmacólogos (A-III).
INFECCIÓN POR EL VIH-2.
El VIH-2 presenta resistencia intrínseca a los ITINN y una sensibilidad variable frente a los IP, siendo DRV el más activo19. Los INI son activos frente al VIH-219 21
Recomendaciones
- Se aconseja monitorizar clínica y CD4 cada 6-12 meses y, si está disponible, la CVP de VIH-2 (A-III).
- El uso de ITINN está contraindicado en el tratamiento de la infección por VIH-2 (A-I).
- El régimen de TAR de inicio en estos pacientes es la combinación de 2 ITIAN + 1 INI (A-II). Como alternativa a los INI, puede aconsejarse DRV/c (C-II).
- En pacientes con infección dual VIH-1/VIH-2 el TAR debe seguir las recomendaciones del VIH-2 (A-III).
- En situación de FV se recomienda hacer un estudio de resistencias. La interpretación de éstas deberá realizarse con un algoritmo específico para VIH-2 (A-III).
- El inhibidor de la fusión enfuvirtide y el FTV no se recomiendan para el tratamiento de personas con VIH-2 (A-III).
- Para los pacientes con virus multirresistentes, el ibalizumab y el LEN demuestran potencia in vitro contra el VIH-2 (B-III).
Tablas:
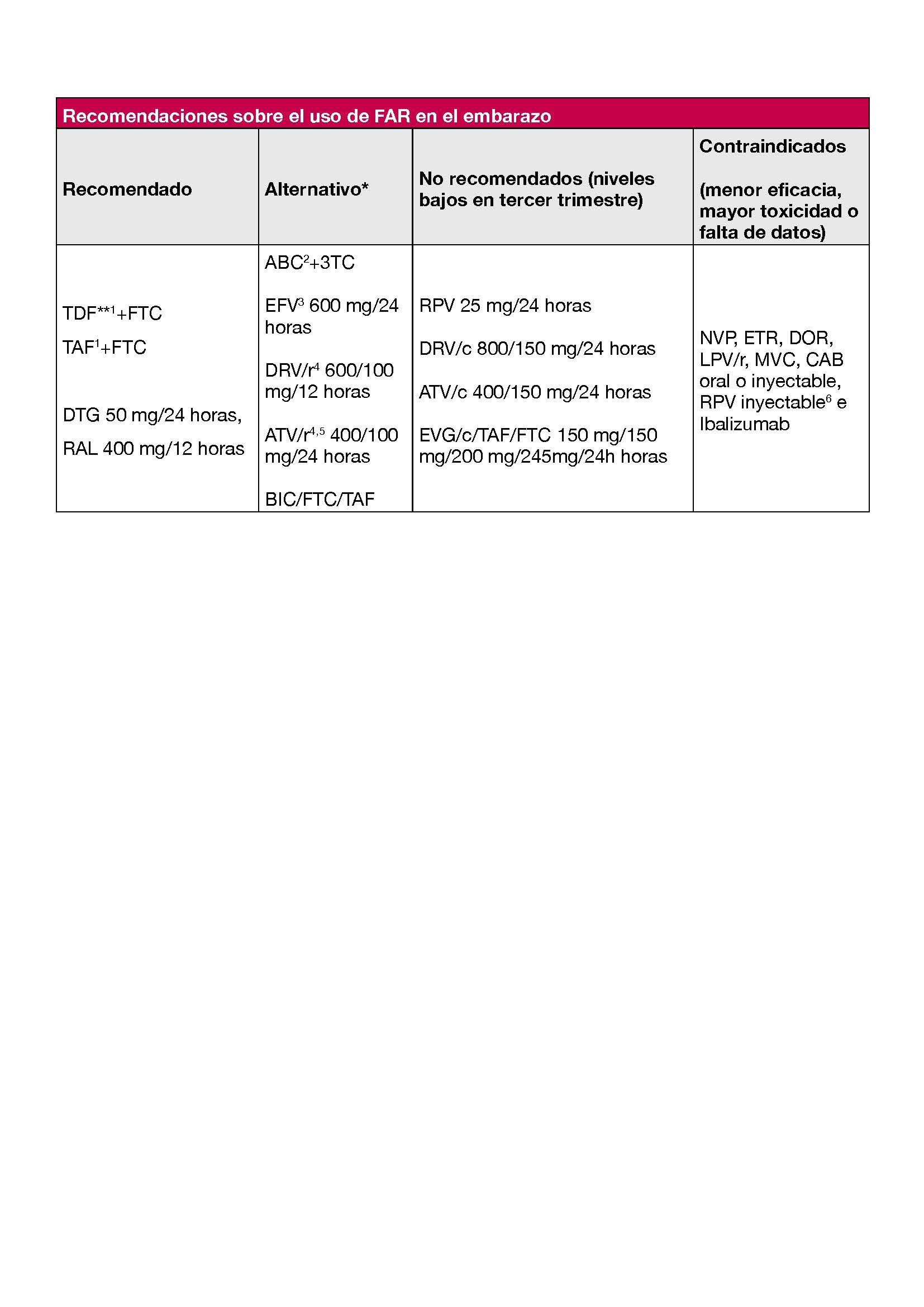
* Fármacos antirretrovirales que se podrían utilizar en sustitución de alguno de los recomendados cuando no puedan utilizarse los fármacos de 1ª elección.
** Con el término de TDF nos referimos a cualquiera de las sales de tenofovir disponibles (fumarato, fosfato, maleato, succinato).
- Debe considerarse especialmente para el tratamiento en pacientes con coinfección por VHB.
- Si el alelo HLA-B*5701 es negativo, aunque con pequeño riesgo de hipersensibilidad.
- Potencialmente teratógeno, aunque datos observacionales sugieren que no hay mayor riesgo de malformaciones congénitas. No es de elección en las 8 primeras semanas de gestación. Si la mujer está tomando ya EFV cuando queda embarazada, es posible continuar su uso.
- Hay evidencia de que ATV/r es igual de efectivo que DRV/r en embrazadas y se tolera mejor.
- Cuando se acompaña de TDF en lugar de ZDV, es imprescindible utilizar la dosis de 400/100 mg en el segundo y tercer trimestre, dada la interacción existente entre TDF y ATV y las especificidades farmacocinéticas de la gestación.
- Cabotegravir oral, y la combinación de formulaciones de acción prolongada de administración im con cabotegravir y rilpivirina no se recomiendan durante el embarazo; carecemos de suficientes datos para recomendar la utilización en pacientes con deseo gestacional. Las mujeres con tratamiento inyectable que se queden embarazadas deben cambiar preferiblemente a un tratamiento oral a las 4 semanas tras la última dosis im.
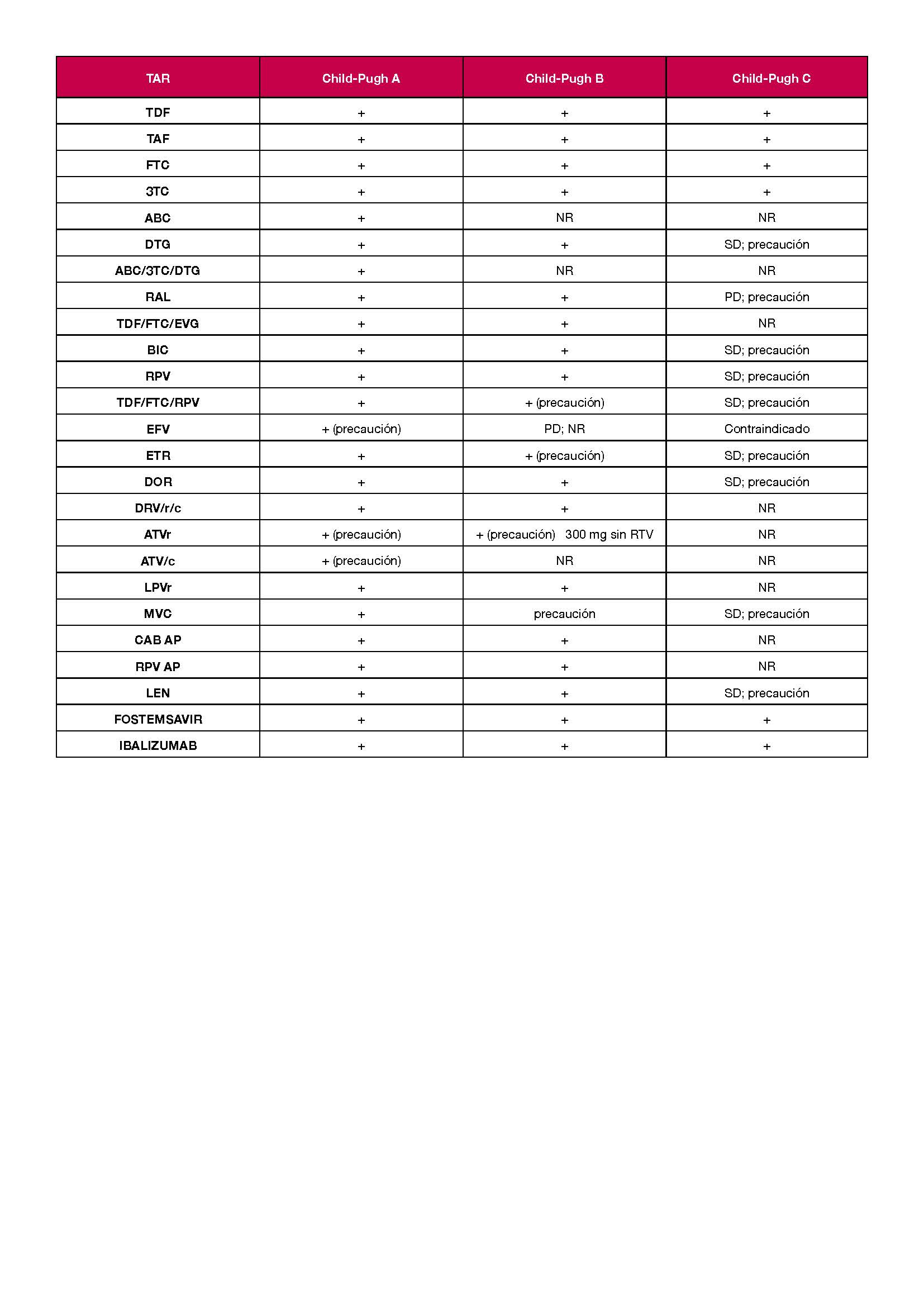
Fármaco seguro, no precisa ajuste de dosis; PD: Pocos datos; SD: sin datos suficientes sobre la dosis recomendable o la seguridad; NR: no recomendado
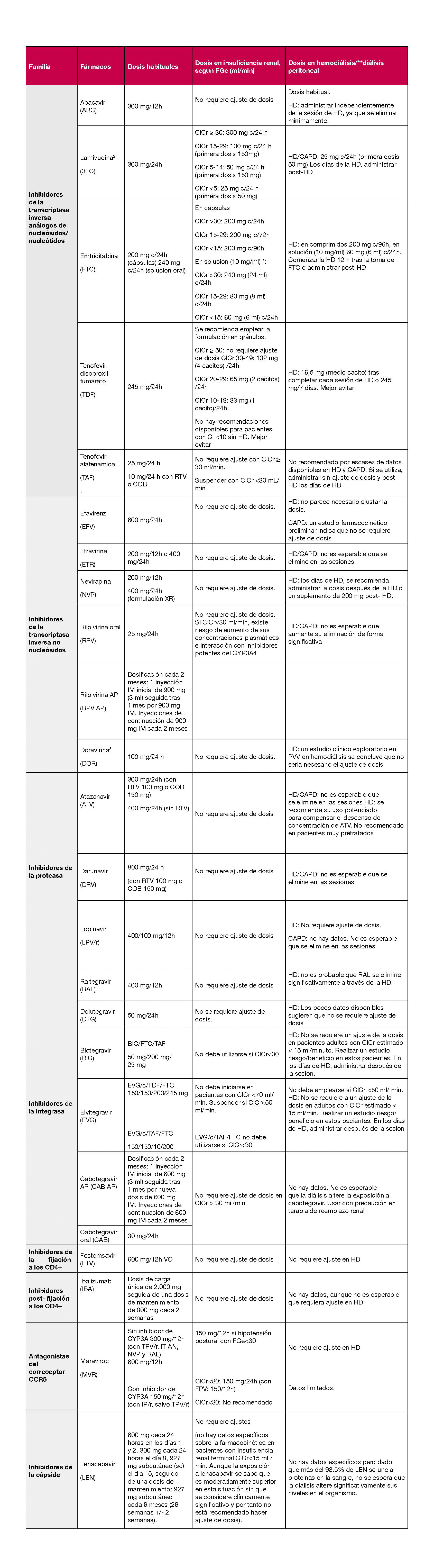
Bibliografía:
Recommendations for the use of antiretroviral drugs in pregnant women with HIV-1 infection and interventions to reduce perinatal HIV transmission in the United States. Departament of health and human services (HHS). Panel on treatment of pregnant women with HIV infection and prevention of perinatal transmission. January 31, 2024. Updated. Accesible en https://clinicalinfo.hiv.gov/en/guidelines/perinatal/whats-new-guidelines.
Documento de consenso para el seguimiento de la infección por el VIH en relación con la reproducción, embarazo, parto y profilaxis de la transmisión vertical del niño expuesto. Grupo de expertos de la División de control de VIH, ITS, Hepatitis virales y Tuberculosis (DCVIHT), Sociedad Española de Ginecología y Obstetricia (SEGO), Grupo de Estudio de Sida (GeSIDA) y Sociedad Española de Infectología Pediátrica (SEIP). Diciembre de 2023. Accesible en https://guiasclinicas.gesida-seimc.org.
Documento de Prevención y Tratamiento de Infecciones Oportunistas y otras Coinfecciones en Pacientes con Infección por VIH. Versión 1.0. Grupo de Estudio del SIDA-SEIMC (GeSIDA). Febrero 2022. Disponible en: https://gesida-seimc.org/wp-content/uploads/2022/03/GUIA_PREVENCION_INFECCIONES_OPORTUNISTAS.pdf
Cattaneo D, Gervasoni C. Dolutegravir dosing with rifampicin. Lancet HIV. 2023 Oct;10(10):e635-e636.
Naidoo A. Center for the AIDS Programme of Research in South Africa, Durban South Africa. Efficacy, safety, and PK of BIC/FTC/TAF in adults with HIV and tuberculosis on rifampicin at Week 24. [Oral abstract Session-14]. CROI 2024. Conference on Retroviruses and Opportunistic Infections. March 3-6, 2024. Denver, Colorado, EE.UU.
Lenacapavir (Sunlenca®). Monografía del producto. Laboratorio Gilead Sciences Ireland UK.
Guía GESIDA/GEHEP del manejo de la enfermedad hepática en el paciente VIHhttps://gesida-seimc.org/wp-content/uploads/2022/11/DocumentoDeConsensoGESIDA-GEHEPSobreElManejoDeLaEnfermedadHepaticaEnElPacienteVIH.pdf
Avihingsanon A, Lu H, Leong CL, et al. Bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir, emtricitabine, and tenofovir disoproxil fumarate for initial treatment of HIV-1 and hepatitis B coinfection (ALLIANCE): a double-blind, multicentre, randomised controlled, phase 3 non-inferiority trial. Lancet HIV. 2023 Oct;10(10):e640-652.
Navarro J. HIV and liver disease. AIDS Rev 2022; 25:87-96.
Van Welzen B, Mudrikova T, El Idrissi A, et al. A review of Non-Alcoholic fatty liver disease in HIV-infected patients: the next big thing? Infect Dis Ther 2019; 8:33-50.
Panel de expertos de Gesida. Documento de consenso de GESIDA para la evaluación y el tratamiento de las enfermedades renales en pacientes con infección por el virus de la inmunodeficiencia humana. (Actualización marzo 2020). http://gesidaseimc.org/wp-content/uploads/2020/07/GUIA_GESIDA_Renal_2020.pdf
Moltó J, Graterol F, Curran A, et al. Removal of doravirine by haemodialysis in people living with HIV with end-stage renal disease. J Antimicrob Chemother 2022; 77:1989- 91.
Paik J. Lenacapavir: First Approval. Drugs. 2022 Sep;82(14);1499-1504. https://doi.org/10.1007/s40265-022-01786-0
Weber EJ, Graham H, West SK, et al. Pharmacokinetics of Lenacapavir in Participants with Severe Renal Impairment. [poster]. CROI 2022, February 12–16, 2022. #434.
Lepik KJ, Wang L, Harris M, et al. Evolving patterns of antiretroviral drug interactions in people with HIV in British Columbia, Canada. AIDS 2022; 36:1105-15.
Deutschmann E, Bucher HC, Jaeckel S, et al. Prevalence of potential drug-drug interactions in patients of the Swiss HIV Cohort Study in the era of HIV integrase inhibitors. Clin Infect Dis 2021; 73: e2145-e2152.
López-Centeno B, Badenes-Olmedo C, Mataix-Sanjuan Á, et al. Polypharmacy and drug-drug interactions in people living with human immunodeficiency virus in the region of Madrid, Spain: A Population-Based Study. Clin Infect Dis 2020; 71:353-62.
NCCN Clinical Practice Guidelines in Oncology. Cancer in People with HIV, Version 2.2024. Disponible en: https://www.nccn.org/professionals/physician_gls/pdf/hiv.pdf.
Tzou PL, Descamps D, Rhee SY, et al. Expanded Spectrum of Antiretroviral-selected Mutations in HIV-2. J Infect Dis 2020; 221: 1962-72.
Smith R, Wu V, Song J, et al. Spectrum activity of raltegravir and dolutegravir against novel treatment-associated mutations in HIV-2 integrase: a phenotypic analysis using an expanded panel of site-direct mutants. J Infect Dis 2022; 226: 497-509.
Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIVS. Department of Health and Human Services. Disponible en: https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv.